Topic outline
-
Desarrollo de scripts y uso de librerías para secuencias biológicas
-
2.3. Desarrollo de scripts y uso de librerías para secuencias biológicas
2.3.1. Desarrollo de scripts y uso de librerías en R
En biología computacional debe estar familiarizado con bibliotecas clave de R para bioinformática y su aplicación en el manejo de secuencias, para ello debe conocer las estructuras de programación en R, así como las librerías que permiten el análisis de secuencias biológicas.
En este campo el desarrollo de scripts en R es una habilidad fundamental, ya que permite automatizar tareas repetitivas, analizar grandes volúmenes de datos biológicos y generar visualizaciones complejas de manera eficiente. Los scripts en R consisten en una serie de comandos organizados que pueden ejecutar procesos como la manipulación de secuencias genómicas, el análisis estadístico de datos experimentales y la interpretación de resultados mediante gráficos y tablas. Esta capacidad de automatización es especialmente útil en bioinformática, donde los investigadores suelen trabajar con grandes datasets provenientes de secuenciación masiva (NGS) o experimentos de microarrays.
El ecosistema de librerías en R es otro pilar esencial para la biología computacional, ya que proporciona herramientas especializadas para el análisis de datos biológicos. Paquetes como Bioconductor, y GenomicRanges permiten realizar tareas específicas como la lectura y manipulación de archivos FASTA, el alineamiento de secuencias y la anotación de regiones genómicas. Por ejemplo, seqinr facilita la extracción y análisis de secuencias de ADN y proteínas, mientras que Bioconductor ofrece un amplio repertorio de herramientas para el análisis de datos genómicos y transcriptómicos. Estas librerías están diseñadas para integrarse de manera eficiente con otros paquetes de R, lo que permite construir flujos de trabajo complejos adaptados a las necesidades de cada proyecto.
Además, el uso de R en biología computacional no se limita al análisis de datos, sino que también abarca la visualización y modelado estadístico. Librerías como ggplot2 permiten crear gráficos de alta calidad para representar patrones en datos biológicos, como la expresión génica diferencial o la distribución de variantes genéticas. Asimismo, los modelos estadísticos implementados en R, como los modelos lineales y mixtos, son esenciales para interpretar los resultados experimentales con rigor científico. La combinación de scripting, análisis y visualización en un entorno unificado convierte a R en una herramienta poderosa para los biólogos computacionales, facilitando la generación de hipótesis, la validación de resultados y la comunicación de hallazgos científicos.
2.3.1.1. Introducción a las principales librerías de bioinformática en R
Como complemento a Bioconductor, revisado previamente, para el manejo de secuencias biológicas se puede usar seqinr, el cual es un paquete de R especializado en bioinformática, diseñado para el manejo, análisis y visualización de secuencias biológicas (ADN, ARN y proteínas). A continuación, se detallan a continuación algunos de sus componentes:
- Acceso a bases de datos biológicas
- Sistema ACNUC: Permite recuperar secuencias directamente de repositorios como GenBank, EMBL o UniProt mediante funciones como query() o getSequence().
- Manejo de archivos
- Formatos compatibles: Importa y exporta archivos en FASTA, GenBank, EMBL y formatos comprimidos (e.g., .gz).
- Análisis estadístico básico
- Contenido de GC: Calcula la proporción de bases Guanina y Citosina con GC(), útil para estudios de estabilidad térmica en ADN.
- Longitud de secuencias: Analiza distribuciones con length() y gráficos de densidad.
- Análisis evolutivo
- Distancias genéticas: Calcula divergencias entre secuencias usando modelos como Tamura-Nei (TN93) o Kimura-2 parámetros (dist.dna() del paquete ape, integrable con seqinr).
- Árboles filogenéticos: Construye árboles con métodos como Neighbor-Joining (requiere paquetes como phangorn o ape para visualización).
- Herramientas para proteínas
- Peso molecular: Usa pmw() para calcularlo sumando los pesos atómicos de los aminoácidos.
- Punto isoeléctrico (pI): Determina el pH donde la proteína tiene carga neutra con pI().
- Visualización
- Gráficos básicos:
- Histogramas de longitud de secuencias.
- Gráficos de barras para contenido de GC o composición de bases.
- Diagramas de sectores para frecuencias de aminoácidos.
Al usar este paquete se debe tener en cuenta que en el manejo de memoria no es eficiente para genomas completos o metagenómicas, donde es mejor usar servidores o herramientas como Biopython.
2.3.2. Uso de scripts en R para el manejo y procesamiento de secuencias biológicas (lectura, filtrado y manipulación de secuencias).
En biología computacional, el uso de scripts en R permite automatizar el análisis de secuencias biológicas (ADN, ARN, proteínas) de manera reproducible y eficiente. A continuación, se detalla su aplicación en tres etapas:
- Lectura de secuencias: Los scripts en R utilizan paquetes como seqinr o Biostrings (de Bioconductor) para importar secuencias desde archivos (FASTA, GenBank) o bases de datos.
- Filtrado de secuencias: Los scripts permiten aplicar criterios para seleccionar subconjuntos de datos:
- Por longitud: Eliminar secuencias demasiado cortas o largas.
- Por calidad o anotaciones: Filtrar usando metadatos (ej: organismos, genes).
- Manipulación de secuencias: Los scripts facilitan operaciones como:
- Extracción de regiones.
- Traducción a proteína.
- Búsqueda de patrones.
- Automatización y flujos de trabajo: Los scripts integran múltiples pasos en un pipeline, como:
- Limpieza: Remover secuencias ambiguas
- Análisis estadístico: Generar tablas resumen
- Exportación: Guardar resultados en formatos estandarizados (FASTA, CSV).
Como ventajas del uso de scripts se puede destacar:
- Reproducibilidad: Los scripts documentan cada paso del análisis.
- Escalabilidad: Manejo de miles de secuencias con modificaciones mínimas.
- Integración: Compatibilidad con herramientas como ggplot2 para visualización o shiny para interfaces interactivas.
Este enfoque es esencial en proyectos de genómica, transcriptómica o estudios evolutivos, donde la estandarización y la eficiencia son críticas.
2.3.3. Ejercicios prácticos sobre el manejo y análisis de archivos FASTA con librerías especializadas.
El formato FASTA es uno de los estándares más utilizados en bioinformática para almacenar secuencias de ADN, ARN y proteínas. Su simplicidad y versatilidad lo convierten en una herramienta fundamental para el intercambio y análisis de datos biológicos. Cada archivo FASTA contiene una o más secuencias, cada una precedida por un encabezado que proporciona información sobre la secuencia, como su identificador o características relevantes. El manejo adecuado de estos archivos es esencial para tareas como el alineamiento de secuencias, la búsqueda de motivos conservados y la anotación funcional de genes. Por ello, es crucial que los profesionales en biología computacional adquieran competencias prácticas en la manipulación de archivos FASTA tanto en BASH, R y Python.
En este conjunto de ejercicios prácticos, nos enfocaremos en el uso de librerías especializadas en R para la lectura, edición y análisis de archivos FASTA. Herramientas como seqinr en R permiten extraer secuencias, realizar filtrados, buscar patrones específicos y preparar los datos para análisis posteriores, como alineamientos múltiples o predicciones estructurales. A través de estos ejercicios, se busca trabajar eficientemente con archivos FASTA, para comprender cómo integrar estos procesos en flujos de trabajo más amplios, facilitando el análisis de datos y la interpretación de resultados biológicos.
Aprende más
Para conocer más sobre R, la importación de archivos y secuencias biológicas y su extración de información, puedes descargar el cuaderno de trabajo ¡Accede aquí!
2.4. Exploración preliminar de datos biológicos en PYTHON
Python, pertenece al grupo de los lenguajes de programación y puede ser clasificado como un lenguaje interpretado, de alto nivel, multiplataforma, de tipado dinámico y multiparadigma. A diferencia de la mayoría de los lenguajes de programación, Python nos provee de reglas de estilos, a fin de poder escribir código fuente más legible y de manera estandarizada. Estas reglas de estilo, son definidas a través de la Python Enhancement Proposal Nº 8 (PEP 8) (Curso Python para Principiantes– Eugenia Bahit, 20212)
Python es un lenguaje de programación creado por Guido van Rossum a principios de los años 90 cuyo nombre está inspirado en el grupo de cómicos ingleses “Monty Python”. Es un lenguaje similar a Perl, pero con una sintaxis muy limpia y que favorece un código legible. Se trata de un lenguaje interpretado o de script, con tipado dinámico,fuertemente tipado, multiplataforma y orientado a objetos. (Python para todos, R. González,2008)
En biología computacional, el análisis de datos biológicos es una tarea fundamental. Python, gracias a su versatilidad y a la gran cantidad de librerías disponibles, se ha convertido en una de las herramientas más populares para la manipulación y exploración de datos biológicos. En esta sección, se abordarán los conceptos básicos para trabajar con datos biológicos en Python, desde la manipulación de secuencias hasta la comparación con otras herramientas como R.
2.4.1. Manipulación y análisis de datos de secuencias en Python.
Dado que python es un lenguaje muy versatil y ampliamente usada, una nutrida comunidad alrededor de el colabora desarrollando herramientas para distintos campos de la ciencia, como referencia puede revisar el libro PYTHON FOR BIOINFORMATICS, de Sebastián Bassi, 2018. Existe una gran oferta de textos en este campo de las ciencias.
La manipulación de secuencias biológicas, como ADN, ARN o proteínas, es una de las tareas fundamentales en bioinformática y biología computacional. Estas secuencias contienen información crucial para entender la estructura, función y evolución de los organismos. Python, gracias a su sintaxis clara y a la gran cantidad de librerías especializadas, se ha convertido en una herramienta indispensable para trabajar con este tipo de datos. Desde la extracción de subsecuencias hasta la de ARN a proteínas, Python ofrece un conjunto de funciones y métodos que permiten realizar operaciones complejas de manera sencilla y eficiente.
En el contexto del análisis de secuencias, Python permite realizar tareas básicas como el conteo de nucleótidos o aminoácidos, la búsqueda de patrones específicos y la transformación de secuencias (por ejemplo, la transcripción de ADN a ARN). Además, con librerías como Biopython, es posible llevar a cabo operaciones más avanzadas, como la alineación de secuencias, la predicción de estructuras proteicas o el análisis filogenético. Estas herramientas no solo facilitan el trabajo con datos biológicos, sino que también permiten automatizar procesos repetitivos, lo que es especialmente útil en proyectos de gran escala.
La manipulación de secuencias en Python no se limita a operaciones básicas; también incluye la lectura y procesamiento de archivos en formatos estándar como FASTA y FASTQ, que son ampliamente utilizados en bioinformática. Estos archivos contienen información esencial sobre las secuencias y, en el caso de FASTQ, datos de calidad asociados. Python permite leer, filtrar y analizar estos archivos de manera eficiente, lo que es crucial para garantizar la precisión y confiabilidad de los resultados. En resumen, Python es una herramienta poderosa y versátil que facilita el análisis y la manipulación de secuencias biológicas, siendo una elección ideal tanto para principiantes como para expertos en el campo de la biología computacional.
Algunas operaciones básicas incluyen:
- Extracción de subsecuencias: Usando slicing (secuencia[inicio:fin]), es posible extraer fragmentos específicos de una secuencia.
- Conteo de nucleótidos o aminoácidos: Mediante funciones como count(), se puede determinar la frecuencia de cada base o residuo.
- Búsqueda de patrones: Con métodos como find() o expresiones regulares (re), se pueden identificar motivos específicos en una secuencia.
- Transformación de secuencias: Por ejemplo, la transcripción de ADN a ARN (T → U) o la traducción de ARN a proteínas.
En Python, las cadenas (o strings) son secuencias inmutables de caracteres utilizadas para representar texto. Se definen mediante comillas simples (' '), dobles (" ") o triples (''' ''' o """ """), y permiten almacenar y manipular datos como palabras, frases o cualquier contenido textual. Las cadenas admiten operaciones como concatenación, slicing (corte), búsqueda, formato y más, además de ser compatibles con métodos integrados para transformaciones, como convertir a mayúsculas, minúsculas o dividir en subcadenas. Su inmutabilidad significa que, al modificarlas, se crea una nueva cadena en lugar de alterar la original.
2.4.2, Lectura y procesamiento de archivos FASTA y FASTQ.
Los archivos FASTA y FASTQ son formatos estándar para almacenar secuencias biológicas. Python permite leer y procesar estos archivos de manera eficiente.
- Archivos FASTA: Contienen secuencias con un identificador que comienza con “>”. Para leerlos, se pueden usar bucles y condicionales para extraer la información relevante.
- Archivos FASTQ: Además de la secuencia, incluyen información de calidad. Estos archivos suelen ser más grandes y requieren un manejo cuidadoso de la memoria.
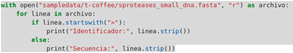
2.4.3. Exploración de datos biológicos usando funciones nativas y librerías básicas de Python.
En Python, los tipos de datos son las categorías que definen cómo se puede manipular y operar con una variable. Algunos tipos comunes incluyen int (números enteros), float (números decimales), bool (valores booleanos: True/False), str (cadena de texto), list, tuple, set, y dict. Entre ellos, las cadenas (strings) son especialmente importantes en bioinformática, ya que se usan para representar secuencias biológicas como DNA, RNA o proteínas. Las cadenas son objetos inmutables que pueden contener texto, y operaciones comunes incluyen concatenación, rebanado y métodos específicos para manipular texto y secuencias. Dada su versatilidad, las cadenas son esenciales en el análisis de datos biológicos, desde la manipulación de secuencias hasta el procesamiento de archivos como FASTA o GenBank.
Aprende más
Para conocer más sobre R, la importación de archivos y secuencias biológicas y su extración de información, puedes descargar el cuaderno de trabajo ¡Accede aquí!
2.4.4. Comparación del manejo de datos entre R y Python.
Tanto R como Python son lenguajes ampliamente utilizados en biología computacional, pero cada uno tiene sus ventajas y desventajas:
Python:
- Sintaxis clara y fácil de aprender.
- Amplio soporte para manipulación de datos y análisis estadístico.
- Librerías especializadas como Biopython, Pandas y NumPy.
- Ideal para integración con otras herramientas y pipelines de análisis.
R:
- Enfoque estadístico más robusto, con librerías específicas para análisis bioinformático (Bioconductor).
- Visualización de datos más avanzada con ggplot2.
- Menos versátil para tareas de programación general o integración con sistemas externos.
Ejemplo comparativo (conteo de nucleótidos):
Python:
secuencia = "ATGCGCTA"
print(secuencia.count("A"))R:
secuencia <- "ATGCGCTA"
print(str_count(secuencia, "A"))La elección entre R y Python dependerá de las necesidades específicas del proyecto y de la familiaridad del usuario con cada lenguaje.
Resumen
La exploración preliminar de datos biológicos en Python es una habilidad esencial para cualquier estudiante de biología computacional. Con las herramientas adecuadas y un buen entendimiento de los conceptos básicos, es posible manipular, analizar y visualizar datos biológicos de manera eficiente. Además, la comparación con R permite entender las fortalezas y debilidades de cada lenguaje, facilitando la elección de la herramienta más adecuada para cada tarea.
En esta sección se profundiza en el uso de R y se cubre una introducción a Python en la biología computacional y a través de las librerías estándar de esta lenguaje trabajando con secuencias biológicas para manipularlas y obtener datos de estas.