Topic outline
-
Estructuras de programación en lenguaje de comandos (BASH)
-
En bioinformática es muy útil para analizar, estudiar, comparar y extraer patrones de cadenas de ADN o ARN y realizar conversiones entre ellas, de tal manera que se facilita el análisis de grandes volúmenes de datos bioinformáticos. Adicionalmente, al desarrollar los scripts de automatización se ahorra tiempo volviendo los procesos de análisis más eficientes o incorporando la posibilidad de calendarizar procesos cuyo tiempo de ejecución, dado el tamaño de los datos, pudiera ser muy extenso limitándose el investigador a ingresar las instrucciones y esperar el resultado durante varias horas o días.
1.1. Preparación del ecosistema bioinformático
Para este curso se requiere de un entorno que tenga los siguientes elementos:
- Terminal BASH
- Entorno de programación para Python ®
- Entorno de programación para R ®
De manera ilustrativa, en caso de que el estudiante no haya tenido experiencias previas con el sistema operativo GNU/Linux®, se han preparado dos máquinas virtuales para facilitar la instalación y configuración de las herramientas necesarias para este curso.
La primera máquina virtual incluye un ecosistema con entorno gráfico denominado Lububio.
La segunda máquina virtual, en cambio, está pensada como un ecosistema de un servidor, el cual no cuenta con entorno gráfico. Sin embargo, el estudiante puede acceder a este recurso a través de un navegador web desde su equipo anfitrión, utilizando el servicio Jupyter® Notebook, que tiene habilitado por defecto los kernel BASH, R® y PYTHON®. A través de las libretas de trabajo en Jupyter®, el estudiante podrá desarrollar y replicar todas las actividades planificadas para este curso.
1.1.1. Lububio como sistema operativo ligero para bioinformática
Lububio está basado en la distribución liviana Lubuntu® y tiene las herramientas necesarias que le permitirán al estudiante conocer de este sistema operativo e interactuar nativamente con su entorno gráfico derivado del cliente de escritorio ®.
1.1.1.1. Introducción a Lubuntu como entorno eficiente para tareas bioinformáticas
Lububio tiene preinstalado un ecosistema que permite realizar tareas dentro del campo de la biología computacional usando el entorno gráfico LXDE® de Lubuntu®. Cuenta con las siguientes herramientas:
- Bash
- Spyder3
- Rstudio
Dado que el objetivo de esta máquina virtual es desarrollar o fortalecer la experiencia en el uso del entorno gráfico liviano LXDE del sistema operativo GNU/Linux®, se recomienda al estudiante seguir las instrucciones de importación y uso básico de esta máquina virtual.
1.1.1.2. Instalación y configuración de herramientas necesarias para manejo de secuencias biológicas.
Mira el Video de Programación Para las Ciencias Biologicas
Revise el video Programación Para las Ciencias Biologicas y realice las siguientes actividades:
- Descargue la imagen Lububio.ova, una vez que la haya descargado, desde ® impórtela como un servicio virtualizado, como se muestra en la Figura 1.
- En la siguiente ventana ubique la imagen Lububio.ova donde la haya guardado en su disco duro, como se presenta en la Figura 2.
- Acepte el acuerdo de licencia, como se muestra en la Figura 3.
- Espere a que termine el proceso de importación, como se indica en la Figura 4.
Aprende más
Descargue la imagen Lububio.ova desde el siguiente enlace ¡Accede aquí!
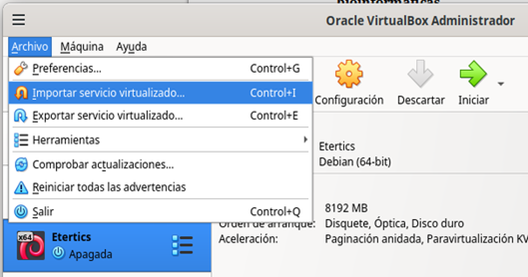
Figura 1: Importación del servicio virtualizado Lububio 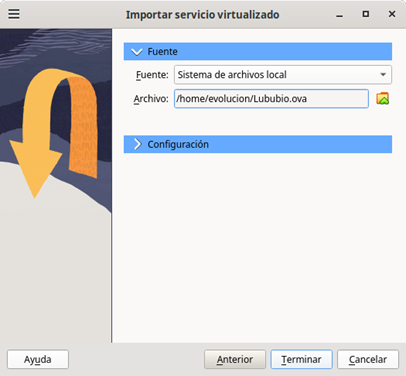
Figura 2: Localización de la imagen Lububio.ova 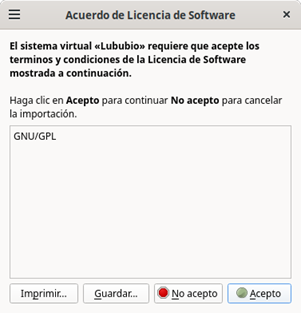
Figura 3: Acuerdo de licencia de Software 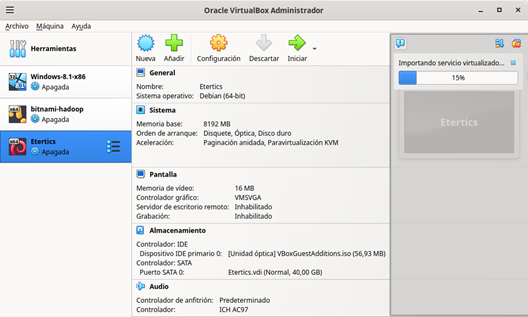
Figura 4: Avance de la importación del servicio virtualizado Lububio El tiempo dependerá de la velocidad y capacidad de su computadora (máquina anfitriona). Cuando el proceso termine deberá tener una nueva máquina virtual llamada Lububio, como se presenta en la Figura 5.
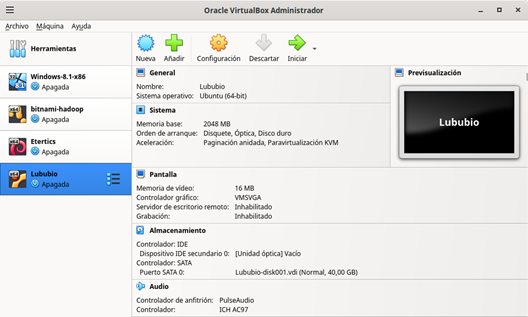
Figura 5: Máquina virtual Lububio Aprende más
Para conocer más sobre el entorno de trabajo en el proceso de importación, puedes ver el siguiente video ¡Accede aquí!
Arranque y Menú de Inicio
- Para arrancar la máquina, hágalo desde Oracle VirtualBox®
- Una vez que se haya iniciado, busque el menú de inicio en la esquina inferior izquierda.
- Pruebe abrir aplicaciones en los 4 escritorios de trabajo.
- Identifique los conceptos “Máquina Anfitriona” y “Máquina Virtual”
- Interactúe con el botón mostrar el escritorio.
Trabajando con la máquina virtual
- Identifique los accesos directos a los tres entornos de trabajo:
- Terminal BASH
- IDE Spyder para Python
- IDE Rstudio para R
- Identifique en el menú de inicio como arrancar los tres entornos de trabajo.
- Identifique las coincidencias de esta máquina con el viejo Bio-Linux:
- Acceda a la documentación de referencia de Bio-Linux
- Identifique que paquetes se podrían instalar en nuestra máquina virtual (sudo apt install nombre_paquete)
- Revise la descripción de algunos de los paquetes de la documentación de referencia de Bio-Linux
- Revise el material introductorio y guía de usuario de Bio-Linux como referencia para nuestra máquina virtual.
- Interactúe con la interfaz gráfica:
- Use el gestor de archivos
- Identifique su carpeta de usuario “manager” y sus subcarpetas
- Identifique el escritorio
- Identifique la papelera
- Recuerde que muchos programas funcionan desde el terminal sin necesidad de un entorno gráfico.
- Interactúe con la interfaz gráfica:
- Cree un archivo en el Escritorio
- Borre el archivo creado
- vacíe la papelera
- Explore el sistema operativo y abra los programas desde el menú de inicio.
- Abra el terminal QTerminal e interactúe con el:
- Lance el navegador Firefox desde el terminal
- Cierre el navegador Firefox interrumpiendo su ejecución
- Familiarícese con el terminal y las rutas del sistema de ficheros
- Pruebe los comandos propuestos en el video.
- Use el terminal QTerminal e interactúe con el:
- Determine la ruta en la en que se encuentre
- Identifique los tipos de ficheros que se pueden listar en QTerminal
- Conozca como regresar de manera sencilla a su carpeta de usuario
- Aprenda a usar el atajo de teclado
- Aprenda a navegar a través del árbol del sistema de ficheros.
Mira el Video de Introducción a comandos Bash y pruebe los comandos propuestos
1.1.2. Configuración de la Máquina Virtual “Servidor Debian 12” con Jupyter y entornos de desarrollo BASH, R y PYTHON
1.1.2.1. Instalación y configuración de Debian 12 como servidor bioinformático.
Mira el Video de Instalacion Debian12 Server Jupyter
Revise este video Instalacion Debian12 Server Jupyter, y realice las siguientes actividades:
- Para importar la imagen OVA, primero descárguela desde este sitio seracademia.com, una vez que la haya descargado, desde Oracle VirtualBox® impórtela como un servicio virtualizado, como se muestra en la Figura 6.
- En la siguiente ventana ubique la imagen Debian12-server-v2.ova donde la haya guardado en su disco duro, como se indica en la Figura 7.
- Espere a que termine el proceso de importación, como se indica en la Figura 8.
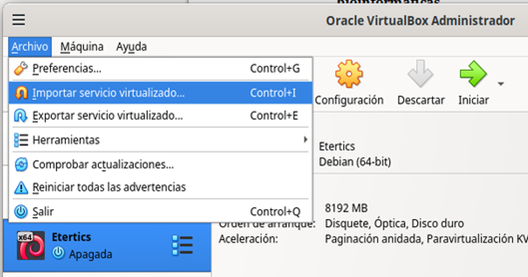
Figura 6: Importación de la imagen ova Debian 12 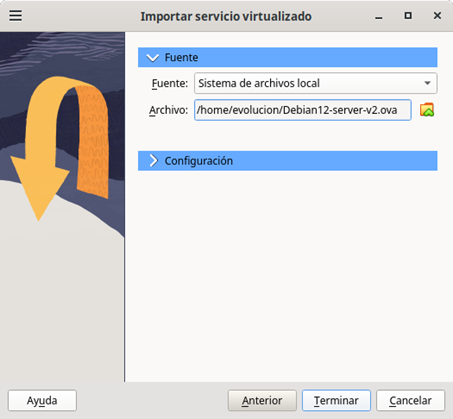
Figura 7: Localización de la imagen Debian 12 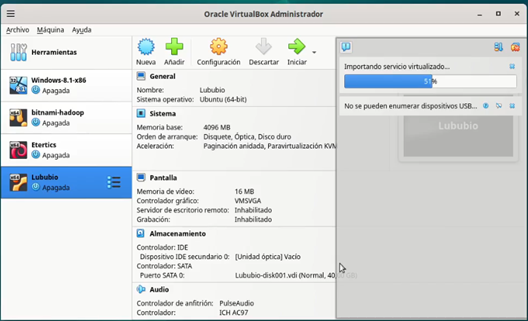
Figura 8: Avance de la importación del servicio virtualizado Debian 12 Cuando el proceso termine deberá tener una nueva máquina virtual llamada Debia12-server, como se muestra en la Figura 9.
Figura 10: Máquina virtual Debian 12 1.1.2.2. Configuración de Jupyter Notebooks en un servidor Debian para el desarrollo de scripts de análisis de secuencias biológicas en BASH
En la máquina virtual con el ecosistema de Debian 12 como servidor al que se le ha retirado el entorno gráfico para ganar en eficiencia y velocidad, se tiene Jupyter previamente configurado con el kernel BASH. Esto permite ejecutar comandos y utilizar herramientas para bioinformática y análisis de secuencias directamente en el ambiente de trabajo de Jupyter a través de un navegador web de la máquina anfitriona.
Este entorno activa la instalación previa del lenguaje de script BASH y proporciona acceso a una variedad de herramientas populares para el análisis de bioinformática, tales como Biopython, Bedtools, Samtools, Pysam, Fastq, y otras herramientas útiles para investigadores y científicos que trabajan en bioinformática ya que proporciona un ambiente completo de análisis sin la necesidad de que el estudiante tenga que instalar herramientas individuales o configurar el sistema operativo.
También permite a los usuarios utilizar comandos en línea y scripts BASH directamente desde el terminal SH y desde este llamar a BASH, o desde la interfaz de Jupyter Notebook para facilitar el trabajo de análisis de bioinformática y la reproducibilidad de los resultados.
Adicionalmente, en este entorno de trabajo de Jupyter Notebook con el kernel BASH en Debian 12, no solo podemos utilizar las herramientas populares para bioinformática mencionadas anteriormente, sino también podemos ejecutar los comandos unix como `tail`, `grep`, `sed` y `awk`. Esto es muy útil en el análisis de datos de bioinformática ya que permite realizar tareas de procesamiento de archivos, filtrado y manipulación de datos rápidamente y de manera eficiente.
Por ejemplo, podemos utilizar `tail` para ver las últimos N líneas de un archivo en un Jupyter Notebook:
1.1.2.3. Instalación de paquetes esenciales para bioinformática en línea de comandos
Para instalar paquetes esenciales para bioinformática en GNU/Linux a través de línea de comandos, se lo puede hacer de dos maneras. La primera, cuando se tiene los programas binarios diseñados específicamente para la distribución, y en el caso de Debian se lo puede hacer usando el gestor de paquetes APT (Advanced Package Tool) que viene preinstalado en la distribución o en su defecto. En caso de que no se cuente con los binarios de la distribución, la segunda manera es a través de compilar los paquetes desde las fuentes del programa que se quiere instalar.
El administrador de paquetes apt, es un utilitario que gestiona los archivos binarios de software compilado y sus dependencias. Hay diferentes administradores de paquetes disponibles para cada distribución de este sistema operativo como, por ejemplo:
- apt (Advanced Package Tool) para sistemas basados en Debian (Ubuntu, Kali Linux, etc.)
- yum (Yellowdog Updater, Modified) para sistemas basados en Red Hat (CentOS, Fedora, RHEL, etc.)
- dnf (Dandified Yum) que es una actualización de yum con mejoras en performance y funcionalidades
- pacman para Arch Linux
- apk para Alpine Linux
El proceso de instalación de paquetes con estos administradores de paquetes generalmente sigue un proceso similar:
- Abrir una terminal y acceder a la cuenta de usuario con privilegios de root o sudo.
- Buscar el paquete deseado utilizando el administrador de paquetes (por ejemplo, apt-cache search nombre_paquete).
- Instalar el paquete utilizando el comando específico del administrador de paquetes.
- Verificar que el paquete se ha instalado correctamente y que está disponible en la carpeta binaria predeterminada (usualmente /usr/bin o /usr/local/bin).
En el cuaderno de trabajo “cuaderno0_BASH.ipynb” disponible en los recursos de la plataforma se desarrollan las actividades que se mencionan a continuación.
Para ilustrar el proceso de instalación se instalarán dos paquetes populares para bioinformática, el primero a través de APT y el segundo compilando el paquete. Como bono y de manera inicial en el cuaderno de trabajo se instalará el paquete cowsay:
Código
sudo apt install cowsay1. Librería Biopython:
Dado que para la librería Biopython se cuenta con los binarios para la distribución Debian®, esta se encuentra disponible en sus repositorios, lo que facilita su instalación con el comando apt.
La biblioteca Biopython es software abierto y multipropósito, escrita en Python, destinada a facilitar el análisis y la manipulación de datos genéticos y bioinformáticos. Biopython proporciona una variedad de utilidades para manipular archivos genéticos comunes (como FASTA, GenBank, SwissProt), así como herramientas para análisis secuencias de ADN, ARN y proteínas, análisis estructural, visualización gráfica y más. También incluye funciones para procesar datos de experimentación, integrar datos externos (como los proporcionados por la base de datos NCBI) y realizar análisis estadísticos y bioinformáticos avanzados.
Para instalar Biopython, ejecute el siguiente comando:
Código
sudo apt-get install python3-biopython2. Bedtools:
Para instalar un paquete desde las fuentes en GNU/Linux utilizando make y make install, primero se necesita descargar los archivos fuente del repositorio git u obtenerlos a partir de un repositorio del desarrollador de la aplicación. Una vez que tenga las fuentes del programa puede seguir los siguientes pasos:
- Descargar las fuentes del repositorio del desarrollador, por ejemplo, git. Para ello debería tener instalado git, en caso de no tenerlo instálelo con apt-get install git.
- Ubíquese dentro de la carpeta que descargó o descomprimió y revise la documentación del desarrollador.
- Instale las dependencias requeridas para compilar el paquete, estas dependencias deberían constar en la documentación del programa. Esto puede ser realizado utilizando apt-get. Dependiendo del sistema operativo. En algunos casos, también es posible instalar las dependencias directamente desde las fuentes si las mismas están disponibles en su repositorio.
- Configurar la instalación utilizando el comando make o configure, dependiendo de cómo lo haya establecido el desarrollador. En algunos casos, esta opción puede estar automatizada mediante una llamada a un archivo setup.sh o configure.sh. Esto configura el paquete para trabajar con el sistema operativo y las dependencias correctas. Para determinar la opción correcta revise detenidamente la documentación del programa.
Como ejemplo ilustrativo se instalará bedtools
Para instalar Bedtools en su última versión desde sus fuentes, podemos utilizar Git para descargar la última versión, instalar las dependencias necesarias y luego ejecutar los scripts de instalación:
Dependencias:
Actualice los repositorios de dependencias
Código
sudo apt updateInstale las dependencias:
Código
sudo apt install build-essential
sudo apt install liblzma-dev
sudo apt install liblz-dev
sudo apt install libbz2-dev
sudo apt install gitDescargar el paquete con GIT:
Código
git clone https://github.com/arq5x/bedtools2.gitCompilar e instalar:
Código
cd bedtools2
make
sudo make installCódigo 1: 1.2. Scripts en línea de comandos (BASH)
Los Scripts en Línea de Comandos (BASH) son una herramienta esencial para la Bioinformática. Estos scripts permiten automatizar tareas repetitivas y personalizar el flujo de trabajo según las necesidades.
En ese sentido se pueden crear scripts para todo tipo de tareas, desde la extracción de datos hasta la visualización de resultados, lo que ayuda a aumentar la productividad y reducir errores humanos.
Adicionalmente, es importante tener en cuenta que los scripts de BASH son muy fáciles de aprender y utilizar, con solo un poco de práctica, se pueden crear scripts eficientes y efectivos que ayuden a simplificar el trabajo en Bioinformática.
Mas adelante en este curso elaborará sus propios scripts con BASH.
1.2.1. Scripting en BASH, variables, control de flujo y estructuras de decisión.
Conceptualmente; scripting en BASH o BASH scripting incluye la creación de scripts que permitan automatizar tareas repetitivas y personalizar el flujo de trabajo según las necesidades, en ese sentido se pueden utilizar variables, control de flujo y estructuras de decisión.
BASH scripting se verá con mayor detalle más adelante. Por ahora y de manera introductoria es importante conocer cómo funcionan y que tipo de variables tenemos en BASH.
Variables
Las variables en BASH son utilizadas para almacenar valores que pueden ser reutilizados dentro del script. Se pueden crear variable asignándoles valores indistintamente de si son números o cadenas de caracteres, ya que en este lenguaje no se requiere declarar previamente las variables y estas toman la naturaleza de lo que se les asigne.
Código
variable1 = "cadena de caracteres"
variable2 = 3El control de flujo permite dirigir el flujo de ejecución de un script según las necesidades, así se puede utilizar condicionales y ciclos para lograrlo. Por ejemplo:
Código
if [ $variable == "valor" ]
then
echo "La variable tiene el valor deseado."
fin if
```Las estructuras de decisión en los scripts permiten tomar diferentes acciones según lo que ocurra durante la ejecución del script, para ello se puede utilizar case, switch o select. Por ejemplo:
Código
case $variable in
"valor1")
echo "Se ha seleccionado valor 1."
;;
"valor2")
echo "Se ha seleccionado valor 2."
;;
*)
echo "Valor no reconocido."
;;
esac1.2.2. Comparación de BASH con otros lenguajes de scripting usados en bioinformática
- Características Generales:
- BASH es un intérprete de comandos de línea de código abierto que se utiliza en Linux y Unix.
- R es un lenguaje de programación estadístico orientado a la ciencia de datos.
- Python es un lenguaje de programación multipropósito, utilizado tanto para ciencia de datos como para desarrollo web.
- Uso en Bioinformática:
- BASH es muy popular en bioinformática debido a su sencillez y flexibilidad. Es fácil de aprender y se puede utilizar para automatizar tareas repetitivas.
- R también es muy utilizado en bioinformática, especialmente para análisis estadísticos. Se trata de un lenguaje muy potente y adaptable que permite trabajar con datos biológicos.
- Python también se utiliza en bioinformática, tanto para el análisis de datos como para la creación de herramientas y paquetes. Es muy flexible y ofrece una gran cantidad de bibliotecas y paquetes para diferentes tareas.
- Ejemplo de uso:
- BASH puede utilizarse para automatizar tareas repetitivas, como la extracción de datos o la visualización de resultados. Por ejemplo, un script BASH podría extraer secuencias de ADN de un archivo BAM y crear un archivo BED.
- R se utiliza especialmente para análisis estadísticos en bioinformática. Por ejemplo, un script R podría analizar datos de expresión génica y crear gráficas que muestren los resultados.
- Python también se utiliza para análisis estadísticos en bioinformática, pero también se puede utilizar para la creación de herramientas y paquetes. Por ejemplo, un script Python podría analizar datos de expresión génica y crear un paquete que permita automatizar el proceso de análisis.
De manera general; BASH es un shell de comandos utilizado en sistemas operativos Unix / Linux que permite a los usuarios ejecutar comandos, redirigir la entrada/salida y crear scripts para automatizar tareas repetitivas. Python es un lenguaje de programación multipropósito utilizado para una variedad de aplicaciones como análisis de datos, Machine Learning, robótica y mucho más. R es un lenguaje estadístico y computacional utilizado principalmente para análisis estadístico y científico.
En sistemas operativos GNU/Linux BASH es una buena opción para ejecutar comandos o automatizar tareas repetitivas, mientras que de manera complementaria Python o R son las mejores opciones para analizar datos o realizar cálculos estadísticos.
Característica BASH Python R Propósito principal Shell scripting, automatización de tareas Desarrollo de software, automatización, ciencia de datos Análisis estadístico y visualización de datos Facilidad de aprendizaje Moderada, basada en comandos de texto Alta, sintaxis clara y legible Moderada, especialmente para estadística Popularidad Alta en administración de sistemas y DevOps Alta en desarrollo general y ciencia de datos Alta en estadística y análisis de datos Sintaxis Sencilla pero precisa, basada en comandos Fácil de leer y escribir, más estructurada Especializada en estadísticas y matrices Ámbito de uso Administración de sistemas, scripts de automatización Desarrollo de aplicaciones, análisis de datos, automatización Análisis de datos, modelado estadístico Bibliotecas y paquetes Limitado en comparación con Python y R Gran cantidad de bibliotecas (como NumPy, pandas, matplotlib) Gran cantidad de paquetes para análisis estadístico Interactividad Limitado a la línea de comandos Interactivo en consolas y notebooks (Jupyter) Interactivo en consolas y notebooks (RStudio) Velocidad de ejecución Muy rápida para tareas simples de administración Moderada, depende de la tarea Moderada, optimizada para estadísticas Soporte para programación orientada a objetos (OOP) No soporta OOP Soporta OOP completamente Limitado soporte para OOP Compatibilidad multiplataforma Sí (Linux, macOS, Windows) Sí (Linux, macOS, Windows) Sí (Linux, macOS, Windows) Comunidad y documentación Amplia comunidad en administración de sistemas Gran comunidad de desarrolladores y científicos de datos Gran comunidad en estadísticas y ciencia de datos Entornos de desarrollo Terminal o editores de texto IDEs como PyCharm, VSCode, Jupyter Notebooks RStudio, Jupyter Notebooks Velocidad de desarrollo Rápido para tareas simples, menos flexible Rápido y flexible para desarrollo general Rápido en tareas estadísticas, menos flexible en desarrollo general Tabla 1: Comparación entre BASH, Python y R Resumen
- Hemos preparado nuestro ecosistema de trabajo importando tanto la máquina virtual con entorno gráfico que permitirá a los estudiantes interactuar con el entorno gráfico LXDE® de GNU/Linux® y la distribución Lubuntu® así como nuestro servidor Debian 12 con Jupyter y los kernel BASH R y Python.
- También hemos visto como arrancar nuestras máquinas virtuales, para usar nuestro entorno de trabajo, ya sea a través del escritorio del sistema operativo como desde Jupyter y un navegador desde la máquina anfitriona.
- Instalamos nuevos paquetes en nuestro sistema operativo tanto con apt como compilándolo y vimos nociones básicas de BASH scripting y los tipos de variables de BASH.
- Finalmente, comparamos BASH con Python y R, lenguajes de programación usados ampliamente en biología computacional y que también veremos en este curso.