Diagrama de temas
-
Trabajo con el terminal BASH y secuencias biológicas.
-
1.3. Uso del terminal BASH
El terminal BASH (Bourne Again Shell) es una herramienta poderosa que permite interactuar con el sistema operativo mediante comandos de texto. En bioinformática, el uso eficiente de BASH y sus comandos es esencial para manejar, procesar y analizar grandes volúmenes de datos biológicos. A través del terminal, podemos realizar tareas como la navegación de directorios, manipulación de archivos y análisis de datos de secuencias biológicas sin necesidad de interfaces gráficas. En esta sección, cubriremos los comandos esenciales que todo biólogo computacional debe conocer.
1.3.1. Comandos esenciales para bioinformática
El uso del terminal BASH es fundamental en bioinformática, ya que permite gestionar, procesar y analizar datos biológicos de manera eficiente. A continuación, exploraremos una serie de comandos básicos y herramientas para manejar datos de secuencias biológicas. Estos comandos forman la base de nuestra interacción con los datos y nos permiten trabajar en proyectos de investigación, análisis de genomas, y más.
1.3.1.1 .Práctica y experiencia con comandos básicos del sistema:
1.3.1.1.1. lsb_release, uname -a: Verificación del sistema operativo y kernel.
Antes de comenzar a trabajar con herramientas de bioinformática, es importante conocer el sistema operativo en el que estamos trabajando. Los comandos y nos proporcionan información esencial sobre el sistema y el kernel en uso.
- lsb_release: Este comando nos da información sobre la distribución de Linux instalada, incluyendo la versión y el nombre del sistema operativo.
- uname -a: Este comando nos proporciona información sobre el sistema operativo y el kernel, incluyendo la versión del kernel de Linux y la arquitectura.
- hostname: sin argumentos mostrará el nombre del host actual, mientras que con un argumento específico, permitirá establecer el nuevo nombre del host localmente.
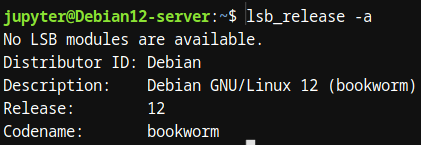

Figura 1: Prueba de Imagen Estos comandos son útiles para conocer las características del sistema antes de ejecutar herramientas bioinformáticas, ya que algunas herramientas pueden depender de versiones específicas del sistema operativo o del kernel.
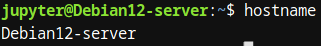
1.3.1.1.2. host, pwd, cd: Navegación en directorios.
La navegación eficiente por los directorios y la manipulación de rutas es esencial cuando se trabaja con grandes volúmenes de datos biológicos. Los comandos pwd, cd y host nos permiten gestionar nuestra ubicación en el sistema de archivos y realizar tareas relacionadas.
- host: Resuelve nombres de dominio a direcciones IP. Este comando puede ser útil para interactuar con servidores de datos o consultar servidores web de bases de datos biológicas.
- pwd: Imprime el directorio de trabajo actual. Este comando es esencial para saber en qué carpeta estamos trabajando en todo momento.
- cd: Cambia el directorio de trabajo. Nos permite movernos entre diferentes carpetas de nuestro sistema de archivos.
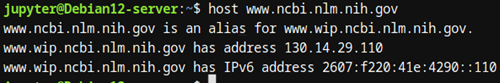
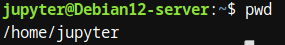
Figura 1: Prueba de Imagen 
Figura 1: Prueba de Imagen Estos comandos nos permiten orientarnos en el sistema de archivos y ubicar los datos que vamos a analizar, facilitando la organización de proyectos de bioinformática.
1.3.1.1.3. mkdir, rmdir, echo, cat, more, less: Manejo básico de archivos.
La manipulación de archivos es una de las tareas más comunes en bioinformática, especialmente al trabajar con datos en formato de texto. Estos comandos nos permiten crear, eliminar, visualizar y manipular archivos.
- mkdir: Crea un nuevo directorio.
- rmdir: Elimina un directorio vacío.
- echo: Imprime un mensaje en la terminal o redirige texto a un archivo.
- cat: Muestra el contenido de un archivo en la terminal. Es muy útil para ver el contenido de archivos de texto y secuencias biológicas.
Código
mkdir datosCódigo
rmdir datosCódigo
echo "ATGCGTAC" > secuencia.txtCódigo
cat sampledata/t-coffee/sample_dnaseq1.fastaSalida
>seq1
GTGAACCTGCGGAAGGATCATTATCGAGTCCAATCCAGCTCATTTCTGGCTGTGCATTCACTTGCACGCCGGGGCGAGTT
GCTCTTGAACTTCTCACACCTGTGCACCTTCTCGTGCCTTCTCCGTCGTCGTCCTTTGTTGGGTGACTCCACCGCTCGC
>seq2
CATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTATCGAAGAGCAACTCTTC
TACACCCTGTGAACTGTCGTCTTAGGACGCAATATAAACTCTAGTGTTGTCAATGAAAGTGTTATACCATAACAAGCAT
>seq3
TCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTATTGAAACTTCTAAGTTT
ACACACCTGTGAACCTGTACATTCCCTTCGGGGTCTTACACACAAACTCTAGTGTCGTCTATGTATGTCTAGTTATAT
- more y less: Permiten visualizar el contenido de archivos grandes de manera paginada, lo que es útil cuando trabajamos con archivos de secuencias de ADN/ARN extensas.
Código
less sampledata/t-coffee/sproteases_large.fasta
o
more sampledata/t-coffee/sproteases_large.fastaEsto le permitirá visualizar el contenido de este archivo y navegador en el con las teclas “flecha arriba” y “flecha abajo” del teclado. Para salir de less o more, presione la letra Q o “Ctrl+c”
Estos comandos nos ayudan a crear la estructura de directorios necesaria para organizar nuestros datos y permiten una fácil visualización de archivos de texto, como las secuencias biológicas.
1.3.1.1.4. grep, wc, cut, sort, awk: Manipulación de texto y análisis de archivos biológicos.
Los comandos de procesamiento de texto son fundamentales cuando se trabaja con archivos de secuencias biológicas, ya que nos permiten buscar, contar, extraer y ordenar datos de manera eficiente.
- grep: Permite buscar patrones de texto dentro de un archivo. Es ideal para encontrar secuencias o características específicas dentro de grandes archivos de secuencias biológicas.
- wc: Cuenta las líneas, palabras y caracteres en un archivo. Es útil para obtener estadísticas rápidas sobre archivos de secuencias.
- cut: Extrae columnas específicas de un archivo de texto, como archivos CSV o tabulados. Es útil para extraer información de archivos que contienen múltiples campos.
- sort: Ordena las líneas de un archivo. Puede ser usado para ordenar secuencias de manera alfabética o numérica.
- awk: Un lenguaje de programación para procesar texto, que permite realizar filtrado, extracción y análisis avanzados. Es especialmente útil para trabajar con archivos en formatos como GFF o TSV.
Código
grep "ATG" sampledata/t-coffee/sproteases_large.fastaCódigo
wc -l sampledata/t-coffee/sproteases_large.fasta
//muestra el número de líneas de ese archivoCódigo 1: Código
cut -d " " -f1 sampledata/t-coffee/struc1.pdb
muestra el primer campo de ese archivo, usando como delimitador de campos el carácter espacio " "Código 1: Código
sort sampledata/t-coffee/struc1.pdbCódigo
awk '{print $1, $3}' sampledata/t-coffee/struc1.pdbEstos comandos de manipulación de texto nos permiten analizar, transformar y filtrar grandes conjuntos de datos biológicos de manera eficiente.
1.3.1.1.5. Prácticas enfocadas en trabajar con archivos de datos biológicos, como secuencias de ADN y ARN.
El objetivo principal de esta sección es que los estudiantes adquieran la habilidad para trabajar con archivos de secuencias biológicas utilizando los comandos mencionados. En la práctica del cuaderno de trabajo “Cuaderno1_BASH_Apellido_Nombre.ipynb”, los estudiantes utilizarán, entre otros, grep para buscar subsecuencias específicas dentro de archivos FASTA, wc para contar las secuencias en un archivo de texto, y tr o fold para extraer y filtrar información de archivos.
Durante estas prácticas, se trabajará con archivos de secuencias biológicas reales, y los estudiantes aprenderán a aplicar comandos de BASH para realizar tareas comunes de bioinformática, como la búsqueda de patrones, la manipulación de datos de secuencias, y la organización de grandes conjuntos de datos.
1.4. Uso avanzado del terminal BASH
En bioinformática, el manejo y análisis de grandes volúmenes de datos biológicos requiere de herramientas y técnicas avanzadas que nos permitan procesar secuencias de manera eficiente. El terminal BASH es una de las herramientas más poderosas para realizar este tipo de tareas. En esta sección, exploraremos cómo usar tuberías, redirección y herramientas como AWK y bioawk para realizar procesamiento de datos biológicos.
1.4.1. Tuberías, filtros y procesamiento de datos biológicos
Tuberías (|): En BASH, la tubería permite conectar dos comandos de tal forma que la salida del primer comando se convierte en la entrada del siguiente. Esta técnica es útil para encadenar varias operaciones y crear flujos de trabajo complejos y eficientes.
• Ejemplo: Supongamos que tenemos un archivo de secuencias FASTA y queremos contar cuántas secuencias contienen un patrón específico, como "ATG". Podemos usar grep para buscar el patrón y luego contar las coincidencias con wc -l:
Código
grep "ATG" sampledata/t-coffee/sproteases_large.fasta | wc -lEn este caso, grep busca todas las líneas que contienen "ATG" en el archivo sampledata/t-coffee/sproteases_large.fasta , y luego wc -l cuenta cuántas líneas fueron encontradas.
- Redirección de salida (>): Permite redirigir la salida de un comando a un archivo, en lugar de mostrarla en la terminal. Esto es útil cuando necesitamos guardar los resultados de un análisis o filtrado para su posterior análisis.
Ejemplo: Si queremos guardar las secuencias que contienen un patrón específico en un nuevo archivo, podemos usar:
Código
grep "ATG" sampledata/t-coffee/sproteases_large.fasta > secuencias_atg.fastaEste comando buscará todas las secuencias que contienen "ATG" en sampledata/t-coffee/sproteases_large.fasta y las almacenará en el archivo secuencias_atg.fasta.
Las tuberías y los filtros son conceptos fundamentales en BASH. Nos permiten tomar la salida de un comando y pasarla como entrada a otro comando, creando flujos de trabajo eficientes y fáciles de gestionar. Estas técnicas son esenciales en bioinformática para manejar y procesar datos biológicos de manera eficiente.
Redirección de entrada (<): Permite usar un archivo como entrada para un comando. Esto es útil cuando queremos que un comando procese datos de un archivo directamente, en lugar de escribirlos manualmente.
Ejemplo: Supongamos que tenemos un archivo llamado datos.txt que contiene una lista de secuencias, y queremos procesarlo con awk:
Código
awk '{print $1}' < datos.txtEste comando tomará el contenido del archivo datos.txt y pasará su contenido a awk para imprimir la primera columna de cada línea.
1.4.1.1. Uso de tuberías para concatenar comandos y flujos de datos bioinformáticos.
El uso de tuberías es muy común en bioinformática, especialmente cuando se trabaja con datos grandes o complejos. Los siguientes ejemplos ilustran cómo combinar comandos de BASH para crear flujos de trabajo complejos en el análisis de datos biológicos:
Ejemplo de procesamiento de archivos FASTA: Si tenemos un archivo FASTA y queremos extraer todas las secuencias que comienzan con "ATG", podemos utilizar un flujo de trabajo que combine varios comandos:
Código
grep "^>.*ATG" secuencias.fasta | cut -d" " -f1 > secuencias_con_atg.fastaEn este ejemplo, grep busca las secuencias que empiezan con "ATG" en el archivo FASTA, y luego cut extrae el primer campo de cada línea (que puede ser el identificador de la secuencia). Finalmente, la salida se redirige a un nuevo archivo secuencias_con_atg.fasta.
Ejemplo de procesamiento de archivos de secuencias: Para contar cuántas secuencias tienen una longitud superior a 100 nucleótidos en un archivo FASTA:
Código
grep -v "^>" secuencias.fasta | awk 'length($0) > 100' | wc -lEste flujo de trabajo usa grep para eliminar las líneas de encabezado (que comienzan con ">"), luego usa awk para filtrar las secuencias cuya longitud es mayor a 100 nucleótidos, y finalmente, wc -l cuenta cuántas secuencias cumplen con esa condición.
1.4.2. Procesamiento de datos biológicos con AWK y herramientas de línea de comandos
AWK es una herramienta extremadamente poderosa para el procesamiento de texto en BASH. Es particularmente útil cuando se trabaja con archivos de texto grandes o datos tabulados, como archivos GFF, VCF, o TSV. AWK permite realizar operaciones complejas de filtrado, extracción, y análisis de datos de manera eficiente.
1.4.2.1. Manipulación de secuencias biológicas usando AWK para extraer, filtrar y analizar datos de forma eficiente.
Extraer columnas específicas de un archivo de datos tabulados como un archivo GFF, que contiene información sobre genes, exones y otras características genómicas.
Código
awk '{print $1, $3, $4, $5}' archivo.gffEste comando imprimirá la primera columna (ID de secuencia), la tercera columna (tipo de característica), y las columnas cuarta y quinta (posiciones de inicio y fin) de cada línea del archivo archivo.gff.
1.4.2.2. Ejemplos prácticos de uso de AWK en archivos de secuencias de ADN/ARN.
Los archivos de secuencias de ADN y ARN, a menudo en formato FASTA, pueden analizarse con AWK de diversas formas:
Contar el número de secuencias en un archivo FASTA:
Código
awk '/^>/ {count++} END {print count}' archivo.fastaEste comando cuenta cuántas secuencias están presentes en el archivo, basándose en el hecho de que cada encabezado de secuencia en FASTA comienza con un ">".
Extraer solo las secuencias de ADN (eliminando encabezados) y concatenarlas en un solo archivo:
Código
awk '!/^>/ {print $0}' archivo.fasta > secuencias_sin_encabezado.fastaEsto elimina las líneas de encabezado (que comienzan con ">") y guarda solo las secuencias de ADN en el archivo de salida.
1.4.2.3. Introducción al uso de bioawk para el análisis de secuencias FASTA y GFF.
bioawk es una versión especializada de AWK, diseñada específicamente para trabajar con archivos biológicos como archivos FASTA, GFF, y VCF. Este programa extiende AWK con funciones adicionales para facilitar el manejo de datos biológicos.
Ejemplo de análisis de secuencias FASTA con bioawk:
Código
bioawk -c fastx '{print $name, $seq}' archivo.fastaEste comando usa el modo fastx de bioawk para extraer y mostrar el nombre y la secuencia de cada entrada en un archivo FASTA.
Ejemplo de análisis de archivos GFF con bioawk:
Código
bioawk -c gff '{print $seqname, $feature, $start, $end}' archivo.gffEste comando usa bioawk para extraer el nombre de la secuencia, el tipo de característica, y las posiciones de inicio y fin de cada línea en un archivo GFF.
Estos comandos de BASH se utilizarán a lo largo del cuaderno de trabajo “Cuaderno2_BASH_Apellido_Nombre.ipynb”. Este cuaderno es una herramienta práctica que permitirá aplicar estos conceptos para trabajar con datos biológicos de manera eficiente utilizando el terminal BASH.
Resumen
En esta clase, se abordaron los conceptos fundamentales de BASH, el terminal de línea de comandos de Linux, y su aplicación en el análisis de datos biológicos. Se cubrió una variedad de herramientas y comandos esenciales para la bioinformática que permiten a los estudiantes trabajar de manera eficiente con secuencias biológicas, como ADN, ARN y proteínas.
- Uso del terminal BASH: Comenzamos aprendiendo sobre los comandos del sistema que permiten navegar por el sistema operativo y gestionar archivos. Comandos como pwd, cd, lsb_release y uname -a fueron utilizados para explorar el entorno de trabajo, verificar el sistema y cambiar entre directorios. Además, se practicaron comandos como mkdir, rmdir, echo, cat, more, y less para manejar y visualizar archivos.
- Manipulación de datos biológicos: Se introdujeron herramientas como grep, wc, cut, sort y awk que permiten manipular archivos de texto, extraer información relevante de secuencias biológicas y realizar análisis estadísticos simples. El uso de estos comandos facilita la filtración, ordenación y procesamiento de datos genómicos y biológicos.
- Uso avanzado del terminal BASH: La clase también cubrió el uso de técnicas más avanzadas como tuberías y redireccionamiento. Estas herramientas permiten encadenar comandos y gestionar el flujo de datos entre ellos, lo que es esencial para crear flujos de trabajo eficientes en bioinformática. Se explicó cómo usar tuberías (|) para pasar la salida de un comando como entrada para otro y cómo redirigir la salida o la entrada de datos a archivos.
- Procesamiento con AWK y bioawk: Se profundizó en el uso de AWK, una herramienta poderosa para procesar y manipular secuencias biológicas, y bioawk, una versión especializada de AWK que está optimizada para trabajar con formatos de datos biológicos como FASTA y similares. Se conoció como extraer, filtrar y analizar datos de secuencias de manera eficiente utilizando estas herramientas.