Diagrama de temas
-
Trabajo con el terminal BASH y secuencias biológicas
-
1.5. Manejo de Archivos de Secuencias Biológicas desde el Terminal
El manejo de archivos de secuencias biológicas en bioinformática es fundamental para el análisis de datos genómicos. Los formatos más comunes incluyen FASTA, FASTQ, GFF y otros formatos específicos. A continuación, se presentan técnicas para manipular estos archivos desde la terminal usando herramientas de Linux y .
1.5.1. Comandos Avanzados para Manipulación de Archivos Grandes de Secuencias
Los archivos de secuencias biológicas, especialmente cuando contienen grandes volúmenes de datos (por ejemplo, resultados de secuenciación de alto rendimiento), pueden ser bastante grandes y difíciles de manejar. Sin embargo, con los comandos adecuados desde BASH se pueden gestionar y procesar de manera eficiente.
1.5.1.1. Lectura y Modificación de Archivos en Formato FASTA, FASTQ, GFF y Otros Formatos Bioinformáticos
El siguiente ejemplo muestra las tres primeras líneas de cada secuencia del archivo FASTA analizado:
Resultado
grep ">" -A3 sampledata/t-coffee/sproteases_small_dna.fasta |head -n20
>sp|P29786|TRY3_AEDAE
atgaaccagttcttgtttgtatcattttgtgctcttttagattccgcgaa
agtgagcgccgccaccttaagtagtggtcgaatcgtaggggggtttcaga
tagatattgctgaagtaccacaccaagtgagcctacagaggtctggtcgt
--
>sp|P35037|TRY3_ANOGA
atgatttctaacaagatagctattctactggcggttttagttgtagctgt
cgcatgtgcacaggctcgagtagcacagcaacaccgcagcgttcaggctt
tacctcgttttctccctcgtcccaagtatgatgtgggtcatcgcatcgtt
--
>sp|P03953|CFAD_MOUSE
atgcattcgagtgtctattttgtggcgttggtgatacttggggccgctgt
atgcgccgcacaacctcgtggaagaattctgggaggacaggaggcagctg
cgcacgcaaggccctatatggcttctgttcaggtaaatggaactcacgtt
--
>sp|P20160|CAP7_HUMAN
atgacgaggctcactgtgttagccttactcgctgggttgcttgcgagttc
cagagctggatcatcaccattgctggatattgtcggtggtcgaaaagcac
gtccgaggcagtttcccttcctggcgagcattcaaaatcaaggtcgccat
--
Para visualizar el contenido de un archivo FASTA , como se mencionó previamente, es posible emplear el comando cat:
Código
cat sampledata/t-coffee/sproteases_small_dna.fasta(se muestran solo las primeras líneas)
Resultado
>sp|P29786|TRY3_AEDAE
atgaaccagttcttgtttgtatcattttgtgctcttttagattccgcgaa
agtgagcgccgccaccttaagtagtggtcgaatcgtaggggggtttcaga
tagatattgctgaagtaccacaccaagtgagcctacagaggtctggtcgt
…..
…..
Mientras que, para buscar patrones o secuencias en un archivo FASTA, se puede usar grep:
Resultado
grep -n "atgaacc" sampledata/t-coffee/sproteases_small_dna.fasta
2:atgaaccagttcttgtttgtatcattttgtgctcttttagattccgcgaa
47:acaccagttacgtgtctctataatgaaccgaacaacgtgcaatctgcgta
209:acagcctatcgaagaatgaacccatgaaacaaacattcgagattaaaaaa
La salida del comando indica que se han encontrado coincidencias coincidencias en las líneas 2, 47 y 209
A continuación se presenta un ejemplo de archivo FASTQ:
Resultado
head SRR957824_1.fastq
@SRR957824.1 1/1
ACTATGAGCGAAACGGCATCCTGGCAGCCGAGCGCATCCATTCCTAACTTATTAAAACGCGCGGCGATTATGGCGGAGAACCCTCGTTTCTTTTCCCATCGTGGAGTGCTGGAGGTGGAGACGCCCTGTATGTGCCAGGCGACGGTAACC
+
?????BBBDDDDDDBDFFFFFFFFFHHHICEEEEHBHHIIIIIIIFGHHIIIIHFGHHCEEHEEHBEDFEFEFD>B@#########################################################################
@SRR957824.2 2/1
GATCGGAAGAGCACACGTCTGAACTCCAGTCACAGCGCTATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAAAAAAATCATACTAATATGAGTTCGATATATAAACTGAAAGATCAAACATGAGTTCATCAGTGTACTATATA
+
????????DDDDDDDDGGGGGGIIIIIIHIIHIIHHHHHHHHHIFHHIIHHHHHHIHGHHIIIFGHDHHEHHE#############################################################################
@SRR957824.3 3/1
TGGCTAACCCGGATACCACTTCCGGCGAAATGTGCGTATTATCCACAGATTCATCGTTTAACACGAATTTTCAAAACGGAACAGCTTATGAGTGCAATCGCGCCTGGAATGATCCTCATCGCGTACCTCTGCGGCTCCATTTCCAGTGCC
Este archivo, perteneciente a la Escherichia coli K1, se descarga con:
Código
wget -c ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR957/SRR957824/SRR957824_1.fastq.gzEl análisis de la calidad de las lecturas que contiene el archivo FASTQ puede realizarse con la herramienta FASTQC. Para instalar esta herramienta en el entorno de trabajo, se emplea el siguiente comando:
Código
sudo apt install fastqcLa ejecución de la herramienta se realiza con el comando fastqc acompañado del archivo FASTQ a analizar:
Resultado
fastqc SRR957824_1.fastq.gz
Started analysis of SRR957824_1.fastq.gz
Approx 5% complete for SRR957824_1.fastq.gz
Approx 10% complete for SRR957824_1.fastq.gz
Approx 15% complete for SRR957824_1.fastq.gz
Approx 20% complete for SRR957824_1.fastq.gz
Approx 25% complete for SRR957824_1.fastq.gz
Approx 30% complete for SRR957824_1.fastq.gz
Approx 35% complete for SRR957824_1.fastq.gz
Approx 40% complete for SRR957824_1.fastq.gz
Approx 45% complete for SRR957824_1.fastq.gz
Approx 50% complete for SRR957824_1.fastq.gz
Approx 55% complete for SRR957824_1.fastq.gz
Approx 60% complete for SRR957824_1.fastq.gz
Approx 65% complete for SRR957824_1.fastq.gz
Approx 70% complete for SRR957824_1.fastq.gz
Approx 75% complete for SRR957824_1.fastq.gz
Approx 80% complete for SRR957824_1.fastq.gz
Approx 85% complete for SRR957824_1.fastq.gz
Approx 90% complete for SRR957824_1.fastq.gz
Approx 95% complete for SRR957824_1.fastq.gz
Analysis complete for SRR957824_1.fastq.gz
Esta herramienta genera un informe visual que describe varios aspectos clave de la calidad de las lecturas (reads) en un archivo FASTQ. Aquí se explican las principales secciones del informe:
1. Basic Statistics
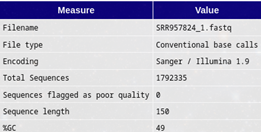
Figura1: fastqc - Basic Statistics Descripción: Resume las características generales del archivo FASTQ.
Incluye:
- Número de secuencias totales.
- Longitud de las secuencias (mínima, máxima y promedio).
- Calidad promedio (Q-score).
- Contenido de GC (%).
2. Per Base Sequence Quality
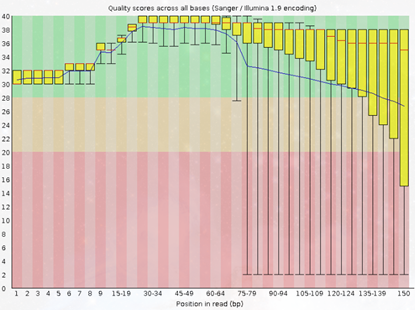
Figura 2: fastqc - Per base sequence quality - Gráfico: Muestra la calidad de las bases en cada posición a lo largo de las lecturas.
- Interpretación:
- Cada caja del gráfico representa la mediana (línea central), el rango intercuartil (caja) y las posiciones extremas (bigotes).
- Alta calidad se refleja en puntajes de calidad (>28) constantes.
Degradación de calidad en las bases finales es común en plataformas como Illumina.
3. Per Sequence Quality Scores
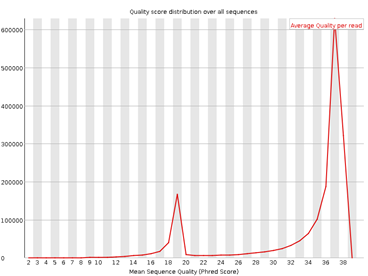
Figura 3: fastqc - Per sequence quality scores - Gráfico: Distribución de las calidades promedio por lectura.
- Interpretación:
- Lecturas con calidades consistentemente altas son ideales.
- Picos en calidades bajas pueden indicar problemas en la secuenciación.
4. Per Base Sequence Content
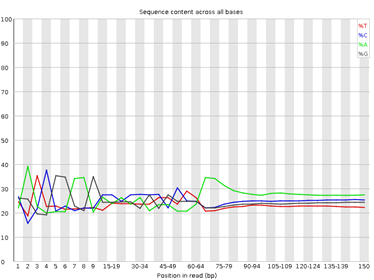
Figura 4: fastqc - Per base sequence content - Gráfico: Proporción de nucleótidos (A, T, C, G) por posición en las lecturas.
- Interpretación:
- En una biblioteca bien balanceada, las proporciones deben ser similares.
- Desequilibrios (especialmente al inicio de las lecturas).
5. Per Sequence GC Content
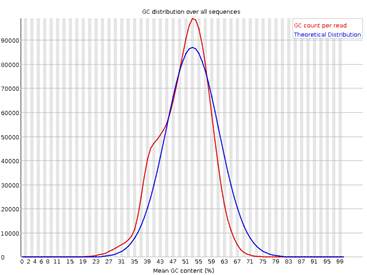
Figura 5: fastqc - Per sequence GC content - Gráfico: Distribución del contenido de GC entre las lecturas.
- Interpretación:
- Un pico único y simétrico es normal.
- Las desviaciones pueden sugerir contaminación por organismos con distinto contenido GC.
6. Per Base N Content
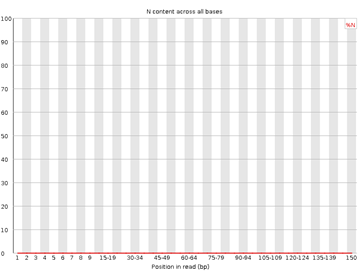
Figura 6: fastqc - Per base N content - Gráfico: Frecuencia de bases no determinadas ("N") por posición.
- Interpretación:
- Idealmente, debe ser cercano a 0.
- Valores altos indican problemas en la calidad de la secuenciación.
7. Sequence Length Distribution
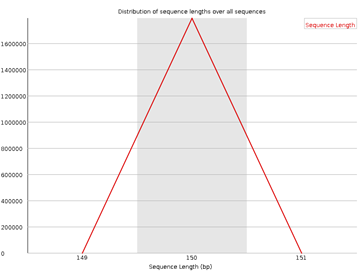
Figura 7: fastqc - Sequence Length Distribution - Gráfico: Longitud de las lecturas.
- Interpretación:
- Una única longitud dominante es típica.
- Variación puede ser esperada en datos de secuenciación adaptativa.
8. Sequence Duplication Levels
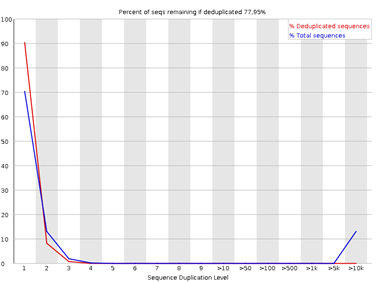
Figura 8: fastqc - Sequence Duplication Levels - Gráfico: Frecuencia de duplicación de lecturas.
- Interpretación:
- Altos niveles de duplicación pueden indicar enriquecimiento intencionado (e.g., RNA-seq) o contaminación.
9. Overrepresented Sequences

Figura 9: fastqc - Overrepresented sequences - Lista: Secuencias presentes en frecuencias anormalmente altas.
- Interpretación:
- Puede indicar contaminantes como adaptadores, primers, o secuencias repetitivas.
10.Adapter Content
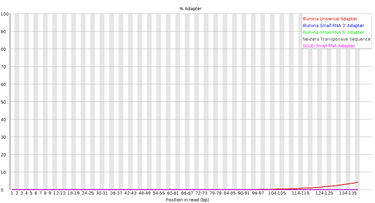
Figura 10: fastqc - Adapter Content - Gráfico: Muestra la proporción de secuencias que contienen adaptadores en cada posición de las lecturas.
- En el eje X: La posición a lo largo de las lecturas.
- En el eje Y: Porcentaje de lecturas con adaptadores detectados.
- Interpretación:
- Idealmente, el porcentaje debe ser cercano a 0%.
- Un aumento hacia el final de las lecturas es común en bibliotecas con fragmentos más cortos que la longitud de lectura.
11. Íconos y su significado:

Figura 11: Significado de íconos - ✔ (Visto verde): Aprobado
- Indica que los datos para esa métrica cumplen con los estándares de calidad esperados.
- No se requieren acciones adicionales.
- Ejemplo: Una buena distribución de calidad por base o un contenido de GC uniforme.
- ! (Advertencia amarilla): Precaución
- Indica que hay algo inusual o ligeramente fuera de los parámetros óptimos.
- Puede no ser un problema grave, pero merece atención para evaluar si afecta el análisis downstream.
- Ejemplo: Ligero desequilibrio en el contenido de bases al inicio de las lecturas.
- ✘ (Cruz roja): Problema detectado
- Indica que hay un problema significativo en esa métrica.
- Puede requerir intervención, como recorte, filtrado, o repetir la preparación/secuenciación de la muestra.
- Ejemplo: Alta proporción de bases de baja calidad o sobre-representación de adaptadores.
13 Interpretación en el contexto del análisis:
- ✔ Verde: Los datos están en buen estado y no necesitas realizar ajustes específicos para esa métrica.
- ! Amarillo: Es una señal para investigar. Si afecta mucho al análisis downstream (como ensamblaje o mapeo), considere tomar medidas correctivas
- ✘ Rojo: Necesita acción inmediata, ya que puede comprometer la utilidad de los datos para análisis posteriores.
Al revisar estos íconos como una guía rápida se puede identificar las áreas que requieren atención en los datos de secuenciación.
Aprende más
Para mayores detalles de A quality control tool for high throughput sequence data revise el siguiente enlace ¡Accede aquí!
GFF (General Feature Format): Los archivos GFF son un estándar ampliamente utilizado en bioinformática para describir características genómicas de secuencias de ADN, ARN o proteínas. Estos archivos contienen información estructurada sobre anotaciones, como genes, exones, intrones, regiones promotoras, sitios de inicio de transcripción, y otros elementos funcionales.
Ejemplo de archivo GFF:
Código
zcat /usr/share/doc/python-biopython-doc/Tests/SwissProt/multi_ex.gff.gzResultado
##gff-version 3
##sequence-region P00750 1 562
P00750 UniProtKB Signal peptide 1 22 . . . .
P00750 UniProtKB Propeptide 23 32 . . . ID=PRO_0000028348
P00750 UniProtKB Propeptide 33 35 . . . ID=PRO_0000028349;Note=Removed by plasmin
P00750 UniProtKB Chain 36 562 . . . ID=PRO_0000028350;Note=Tissue-type plasminogen activator
P00750 UniProtKB Chain 36 310 . . . ID=PRO_0000028351;Note=Tissue-type plasminogen activator chain A
P00750 UniProtKB Chain 311 562 . . . ID=PRO_0000028352;Note=Tissue-type plasminogen activator chain B
P00750 UniProtKB Domain 39 81 . . . Note=Fibronectin type-I
P00750 UniProtKB Domain 82 120 . . . Note=EGF-like
P00750 UniProtKB Domain 127 208 . . . Note=Kringle 1
P00750 UniProtKB Domain 215 296 . . . Note=Kringle 2
Si se desea buscar patrones en un archivo GFF, se puede usar grep, en archivos no comprimidos, y zgrep, en archivos comprimidos en formato .gz:
Código
zgrep "Peptide" /usr/share/doc/python-biopython-doc/Tests/SwissProt/multi_ex.gff.gzResultado
P28799 UniProtKB Peptide 18 47 . . . ID=PRO_0000012694;Note=Paragranulin
P28799 UniProtKB Peptide 58 113 . . . ID=PRO_0000012695;Note=Granulin-1
P28799 UniProtKB Peptide 123 179 . . . ID=PRO_0000012696;Note=Granulin-2
P28799 UniProtKB Peptide 206 261 . . . ID=PRO_0000012697;Note=Granulin-3
P28799 UniProtKB Peptide 281 336 . . . ID=PRO_0000012698;Note=Granulin-4
P28799 UniProtKB Peptide 364 417 . . . ID=PRO_0000012699;Note=Granulin-5
P28799 UniProtKB Peptide 442 496 . . . ID=PRO_0000012700;Note=Granulin-6
P28799 UniProtKB Peptide 518 573 . . . ID=PRO_0000012701;Note=Granulin-7
1.5.1.2. Uso de head, tail y split para la Gestión de Archivos de Secuencias Grandes
Como se vio previamente, cuando trabajamos con archivos de secuencias biológicas grandes, es útil utilizar comandos como head y tail para visualizar el contenido de un archivo sin necesidad de cargarlo completamente en memoria.
split
En ocasiones, cuando un archivo es muy grande se lo puede dividir para realizar análisis más sencillos con los pedazos de archivos más pequeños. Para ello se puede usar el comando split que puede dividir el archivo con base al criterio de líneas o de tamaño de archivo. En biología computacional puede ser conveniente dividirlo por líneas.
De manera ilustrativa trabajaremos en el archivo “SRR957824_1.fastq.gz” que se descargó previamente, inicialmente lo descomprimiremos con gunzip y lo dividiremos en segmentos, cuyos nombres empezaran con “xaa”, “xab”, … etc.
Código
gunzip SRR957824_1.fastq.gzPara saber cuántas líneas tiene originalmente, se puede usar “wc -l” en el archivo descomprimido:
Código
wc -l SRR957824_1.fastqLo que arroja:
Resultado
7169340 SRR957824_1.fastq Es decir, más de 7 millones de líneas, por lo que lo dividiremos en archivos de 200000 líneas cada uno.
Código
split -l 200000 SRR957824_1.fastqEstos archivos nuevos pueden analizarse de manera individual. Por ejemplo, de manera ilustrativa, nuevamente con fastqc.
Código
fastqc xaaResultado
Started analysis of xaa
Approx 5% complete for xaa
Approx 10% complete for xaa
Approx 15% complete for xaa
Approx 20% complete for xaa
Approx 25% complete for xaa
Approx 30% complete for xaa
Approx 35% complete for xaa
Approx 40% complete for xaa
Approx 45% complete for xaa
Approx 50% complete for xaa
Approx 55% complete for xaa
Approx 60% complete for xaa
Approx 65% complete for xaa
Approx 70% complete for xaa
Approx 75% complete for xaa
Approx 80% complete for xaa
Approx 85% complete for xaa
Approx 90% complete for xaa
Approx 95% complete for xaa
Approx 100% complete for xaa
Analysis complete for xaa
1.5.2. Ejercicios Prácticos para Extraer y Procesar Subregiones de Secuencias Biológicas
Para procesar subregiones de secuencias biológicas, por ejemplo, con herramientas como cut, awk y sed se puede extraer ciertas posiciones de una secuencia de ADN o ARN para manipular las cadenas de texto que contienen las secuencias.
Ejemplo: Extraer un fragmento de secuencia de un archivo FASTA
Si se tiene un archivo FASTA y se necesita extraer una subregión de la secuencia (por ejemplo, de la posición 10 a la 50), se puede usar sed o awk.
Con fines ilustrativos, se puede usar sed para extraer la subcadena:
Código
sed -n '2s/\(.\{9\}\)\(.\{41\}\)/\2/p' sampledata/t-coffee/sproteases_small_dna.fastaEste comando extrae 41 bases. Comienza desde la posición 10 de la secuencia de la línea 2 de la primera secuencia delarchivo sproteases_small_dna.fasta.
Esta instrucción:
- Lee la segunda línea del archivo.
- Extrae los caracteres del 10 al 50.
- Imprime únicamente esos caracteres en la salida estándar.
Análisis detallado de la instrucción:
- sed -n: Invoca el comando sed (stream editor) en modo "silencioso". No imprime nada automáticamente, a menos que se indique con el modificador p (print).
- 2s: Aplica la operación de sustitución (s/.../.../) únicamente a la segunda línea del archivo.
- Patrón de búsqueda: \(.\{9\}\)\(.\{41\}\):
- (.{9}): Los primeros 9 caracteres de la línea (grupo 1).
- (.{41}): Los siguientes 41 caracteres de la línea (grupo 2).
- Los paréntesis (...) agrupan estas partes como subexpresiones para referencia posterior.
- Reemplazo: \2:
- Sustituye todo el contenido de la línea con el segundo grupo capturado (los caracteres 10 al 50 de la línea original).
- El primer grupo (\1) es descartado.
- p (print): Imprime la línea modificada después de la sustitución.
Busca una secuencia en la línea que coincida con:
Otro para Extraer y Procesar Subregiones de Secuencias Biológicas será traducir
1.6. Estructuras de Programación en BASH Aplicadas a Bioinformática
En bioinformática, muchas tareas de análisis y procesamiento de secuencias requieren automatización, lo que se puede lograr utilizando estructuras de programación en Bash. Estas estructuras permiten ejecutar operaciones repetitivas, condicionales y de control de flujo, lo que facilita la gestión de grandes volúmenes de datos.
1.6.1. Estructuras de Control: IF, FOR, WHILE
- IF: Se utiliza para realizar decisiones condicionales.
- FOR: Se utiliza para iterar sobre una lista de elementos o secuencias.
- WHILE: Se utiliza para ejecutar un bloque de código mientras se cumpla una condición.
Código
if [ "$secuencia" == "ATG" ]; then
echo "Secuencia encontrada"
fiCódigo
for archivo in *.fasta; do
echo "Procesando $archivo"
doneCódigo
while read línea; do
echo "Leyendo: $línea"
done < archivo.fasta1.6.1.1. Implementación de Ciclos y Estructuras Condicionales para Automatización de Tareas Bioinformáticas
En análisis bioinformáticos, estas estructuras son esenciales para automatizar tareas como el procesamiento de secuencias, la búsqueda de patrones o la conversión de archivos.
Ejemplo de automatización con IF y FOR:
Código
for archivo in *.fastq; do
if grep -q "AGCT" "$archivo"; then
echo "Secuencia encontrada en $archivo"
else
echo "Secuencia no encontrada en $archivo"
fi
done
Este recorre todos los archivos FASTQ en el directorio, buscando una secuencia específica ("AGCT") en cada archivo y proporcionando una salida basada en si la secuencia está presente.
1.6.2. Iteración sobre Secuencias
Cuando se trabaja con múltiples archivos o secuencias, es esencial iterar sobre ellos para realizar análisis masivos o automáticos.
1.6.2.1. Implementación de Bucles para Procesar Múltiples Archivos de Secuencias de ADN o ARN
Ejemplo de bucle sobre archivos de secuencias:
Código
for archivo in sampledata/t-coffee/*.fasta; do
if grep -q "AGCT" "$archivo"; then
echo "Secuencia encontrada en $archivo"
else
echo "Secuencia no encontrada en $archivo"
fi
done
1.6.2.2. Automatización de Tareas Repetitivas para Análisis Masivo de Secuencias
Automatizar tareas repetitivas, como la alineación de secuencias, la extracción de características o la conversión de formatos, es esencial cuando se gestionan grandes volúmenes de datos. Los bucles for y while, junto con el uso de herramientas de bioinformática como BLAST, SAMtools, o BEDTools, hacen que estos análisis sean mucho más eficientes.
Este recorre todos los archivos .fasta e imprime el nombre de cada secuencia.
Código
for archivo in sampledata/t-coffee/*.fasta; do
echo "Procesando el archivo $archivo"
# Realizar procesamiento, por ejemplo, contar nucleótidos
bioawk -c fastx '{print $name}' "$archivo"
done
Conexiones remotas con BASH
En BASH, las conexiones remotas pueden ser establecidas utilizando el protocolo SSH (Secure Shell), el cual es un protocolo de comunicación que permite a los usuarios conectarse a servidores remotos y ejecutar comandos en el servidor. El protocolo utiliza algoritmos de cifrado avanzados para asegurar la privacidad y seguridad de la información transmitida durante la conexión.
Las conexiones remotas con SSH se pueden establecer utilizando el terminal de GNU/Linux, la consola CMD de Windows® o programas de terceros como PuTTY®. Este comando usa la siguiente sintaxis:
Código
ssh [usuario]@[dirección]Donde:
- usuario: es el nombre del usuario que se desea utilizar para acceder al servidor.
- dirección: la dirección IP o el nombre de dominio del servidor.
Por ejemplo, si desea conectarse como usuario jupyter a un servidor con la dirección IP 192.168.1.100, puede utilizar la siguiente sintaxis:
Código
ssh root@192.168.1.100Una vez que se ha establecido la conexión SSH, puede comenzar a ejecutar comandos en el servidor. La conexión se mantiene durante la sesión de shell, y puede cerrarla utilizando el comando exit.
El puerto de conexión por lo general es el 22; sin embargo esto puede ser definido al configurar este servicio en el servidor.
En nuestro caso, la máquina virtual que hospeda la máquina anfitriona escucha las conexiones SSH en el puerto 2224 y lo direcciona internamente al 22.
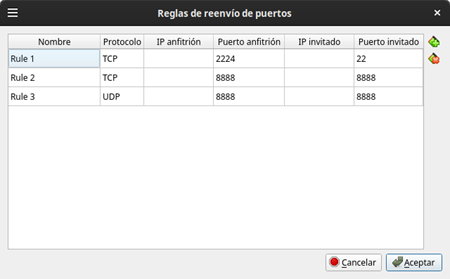
Figura 12: Redireccionamiento en Virtualbox® del puerto 2224 al 22 De manera ilustrativa puede conectarse desde windows usando el programa Putty [https://www.putty.org/] e ingresando los parámetros de conexión como se muestra en la Figura 11. Nótese el usuario localhost y el puerto 2224:
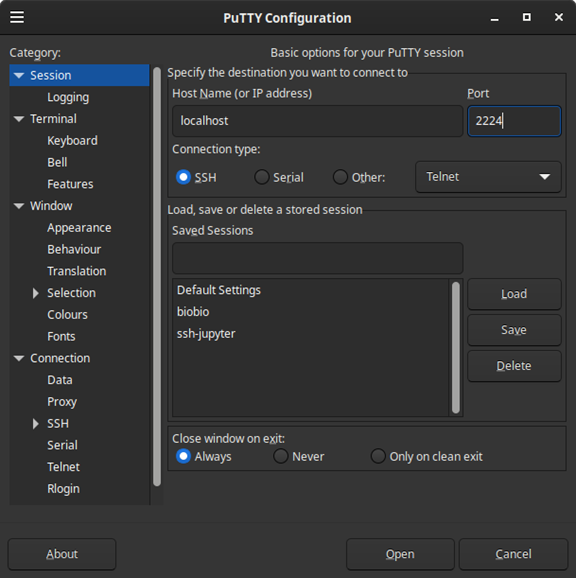
Figura 13: Programa Putty para conexiones remotas Al ejecutarlo se desplegará la ventana de conexión que solicita el usuario y contraseña
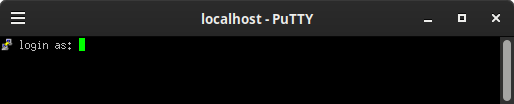
Figura 14: Ventana de conexión remota del programa Putty 
Figura 15: Ventana de conexión remota con los datos ingresados 
Figura 16: Ventana remota del servidor accedido a través de Putty En resumen, las conexiones remotas con SSH permiten a los usuarios conectarse a servidores remotos y ejecutar comandos en el servidor de forma segura. Conocer cómo se establecen estas conexiones es fundamental para cualquier usuario que trabaje en entornos de línea de comandos.
Resumen
En este documento hemos explorado diversas herramientas y técnicas para el manejo eficiente de archivos de secuencias biológicas desde la línea de comandos. Se han abordado comandos avanzados para la manipulación de archivos de gran tamaño en formatos ampliamente utilizados en bioinformática, como FASTA, FASTQ y GFF, además del uso de herramientas como head, tail y split para facilitar la gestión de estos datos. Asimismo, se presentaron ejercicios prácticos enfocados en la extracción y procesamiento de subregiones de secuencias biológicas, permitiendo aplicar los conceptos aprendidos en escenarios reales.
También se ha profundizado en el uso de estructuras de programación en BASH aplicadas a la bioinformática, destacando el uso de estructuras de control como IF, FOR y WHILE para la automatización de tareas. Se han presentado ejemplos prácticos de su implementación en el análisis de archivos de secuencias biológicas, así como la aplicación de bucles para procesar múltiples archivos de ADN o ARN de manera eficiente. Finalmente, se ha abordado la automatización de tareas repetitivas, facilitando el análisis masivo de secuencias y optimizando los flujos de trabajo bioinformáticos.
Adicionalmente, se ha cubierto el proceso de conexión remota a servidores mediante el uso del protocolo SSH, proporcionando las bases para acceder de manera segura a sistemas remotos, ejecutar comandos y gestionar archivos de secuencias biológicas desde entornos de computación de alto rendimiento.