Diagrama de temas
-
Bioinformática con lenguajes de programación multiparadigma
-
2.1 Introducción a la Bioinformática en R y PYTHON
2.1.1 Conceptos básicos de bioinformática aplicados a R y Python.
La bioinformática es una disciplina que integra conocimientos de biología, informática y estadística para analizar y gestionar datos biológicos, como secuencias de ADN, ARN y proteínas. Para trabajar en este campo, es fundamental comprender conceptos básicos como el manejo de secuencias biológicas, el alineamiento de secuencias, la anotación genómica y el análisis de expresión génica. Tanto R como Python ofrecen herramientas y bibliotecas especializadas que facilitan la aplicación de estos conceptos en la práctica. Por ejemplo, en Python, la biblioteca Biopython permite manipular secuencias, realizar búsquedas en bases de datos biológicas y ejecutar algoritmos de alineamiento como BLAST. En R, paquetes como Bioconductor proporcionan funciones para analizar datos genómicos y transcriptómicos, lo que permite a los investigadores explorar patrones y relaciones en grandes conjuntos de datos biológicos.
Uno de los conceptos clave en bioinformática es el alineamiento de secuencias, que consiste en comparar dos o más secuencias para identificar regiones de similitud. Este proceso es esencial para entender la evolución molecular, predecir funciones de genes o identificar mutaciones. En Python, herramientas como Biopython y libraries como PyAlign facilitan la implementación de algoritmos de alineamiento, como Needleman-Wunsch o Smith-Waterman. En R, el paquete Biostrings ofrece funciones para trabajar con secuencias biológicas y realizar alineamientos básicos. Además, ambos lenguajes permiten integrar herramientas externas, como Clustal Omega o MUSCLE, para tareas más complejas. Estos recursos son fundamentales para estudios de filogenia, identificación de genes homólogos o análisis de variantes genéticas.
Otro aspecto importante es el análisis de datos de expresión génica, que permite estudiar cómo los genes se activan o desactivan en diferentes condiciones biológicas. En R, paquetes como DESeq2 y edgeR son ampliamente utilizados para analizar datos de RNA-seq, identificar genes diferencialmente expresados y realizar clustering de patrones de expresión. En Python, bibliotecas como Scanpy y PyDESeq2 ofrecen funcionalidades similares, permitiendo a los investigadores trabajar con datos de expresión génica de manera eficiente. Ambos lenguajes también facilitan la visualización de resultados mediante gráficos interactivos y personalizables, lo que es crucial para interpretar y comunicar hallazgos científicos.
2.1.2 Diferencias clave entre R y Python en el contexto de la bioinformática.
En el contexto de la bioinformática, R y Python son dos lenguajes de programación ampliamente utilizados, cada uno con sus propias fortalezas y enfoques. A continuación, se presentan las diferencias clave entre ambos:
Enfoque y especialización:
R:
Está específicamente diseñado para el análisis estadístico y la visualización de datos, lo que lo hace ideal para tareas bioinformáticas que requieren un enfoque estadístico robusto, como el análisis de expresión génica, la identificación de genes diferencialmente expresados y la creación de gráficos complejos. Paquetes como Bioconductor, DESeq2 y ggplot2 son ejemplos de su especialización en estadística y visualización.
Python:
Es un lenguaje de propósito general con una sintaxis clara y fácil de aprender, lo que lo hace más versátil para una amplia gama de aplicaciones en bioinformática. Python es especialmente fuerte en la automatización de flujos de trabajo, el procesamiento de grandes volúmenes de datos y la implementación de algoritmos de machine learning. Bibliotecas como Biopython, pandas y scikit-learn son ampliamente utilizadas en este contexto.
Ecosistema y bibliotecas:
R:
Cuenta con un ecosistema rico en paquetes especializados para bioinformática, muchos de los cuales están disponibles a través de Bioconductor. Estos paquetes están altamente optimizados para tareas específicas, como el análisis de secuencias, la anotación genómica y la visualización de datos biológicos.
Python:
Tiene un ecosistema extenso y diverso, con bibliotecas que cubren una amplia gama de aplicaciones. En bioinformática, Python ofrece bibliotecas como Biopython para la manipulación de secuencias, Scanpy para el análisis de datos de expresión génica y PyRosetta para el modelado de proteínas. Además, Python es muy utilizado en la integración de herramientas y pipelines de bioinformática así como en aprendizaje automático.
Sintaxis y facilidad de uso:
R:
Tiene una sintaxis que puede ser menos intuitiva para aquellos que no tienen experiencia previa en programación, especialmente en comparación con Python. Sin embargo, su sintaxis está altamente optimizada para operaciones estadísticas y de manipulación de datos, lo que puede resultar en código más conciso para tareas específicas.
Python:
Es conocido por su sintaxis clara y legible, lo que lo hace más accesible para principiantes y para aquellos que provienen de diferentes disciplinas. Esta facilidad de uso, combinada con su versatilidad, hace que Python sea una opción popular para la enseñanza y la implementación rápida de soluciones en bioinformática.
Comunidad y soporte:
R:
Tiene una comunidad muy activa en el ámbito de la estadística y la bioinformática, con una gran cantidad de recursos, tutoriales y foros de discusión disponibles. La comunidad de Bioconductor es particularmente robusta y ofrece un soporte excelente para usuarios de bioinformática.
Python:
También cuenta con una comunidad grande y activa, que abarca una amplia gama de disciplinas, incluyendo la bioinformática. La disponibilidad de recursos y soporte es excelente, y hay una gran cantidad de documentación, tutoriales y foros disponibles para ayudar a los usuarios.
Característica R Python Enfoque principal Análisis estadístico y visualización Procesamiento de datos y modelado computacional Ecosistema biológico Bioconductor, edgeR, DESeq2, Biostrings Biopython, scikit-learn, pandas, BioSimSpace Facilidad de uso Más especializado en estadística, ideal para análisis genómicos Más versátil, ideal para inteligencia artificial y simulaciones Manejo de datos masivos Menos eficiente en grandes volúmenes de datos Excelente para manejar big data y cálculos en paralelo Visualización ggplot2, base R Matplotlib, Seaborn Comunidad y soporte Fuerte en biología estadística y genómica Amplia comunidad en aprendizaje automático y modelado Tabla1: Diferencias entre R y Python en el contexto bioinformático. 2.1.3 Breve introducción a los tipos de datos biológicos comunes (secuencias de ADN, ARN, proteínas) y su representación en ambos lenguajes.
Los tipos de datos biológicos comunes incluyen secuencias de ADN, ARN y proteínas, que se representan en lenguajes como R y Python para el análisis bioinformátiico. En R, los paquetes Bioconductor e Iranges permiten manejar secuencias y intervalos genómicos, mientras que en Python, herramientas como Biopython facilitan la lectura, procesamiento y manipulación de secuencias biológicas. Las proteínas se representan mediante secuencias aminoacídicas o estructuras 3D, usando bibliotecas como NumPy y Pandas para analizar datos estructurados. Estos enfoques permiten realizar desde todo tipo de análisis en biología computacional.
2.1.4 Instalación de entornos de trabajo (Jupyter Notebooks).
Su entorno de trabajo ya está preparado en la máquina virtual Debian 12-server, que fue habilitada al inicio de este curso, sin embargo si requiere instalar jupyter sobre Debian 12®, puede hacerlo a través de:
Instalar las librerías necesarias:
Código
sudo apt install python3 python3-pip python3-venv nodejsCrear un ecosistema python para jupyter:
Código
python3 -m venv jupyterActivar el ecosistema python
Código
source jupyter/bin/activateEsto debería mostrar su promtp con referencia a este ecosistema, por ejemplo:

Figura 1: Activar jupyter Dentro del ecosistema instale jupyter
Código
pip3 install jupyterUna vez instalado puede lanzarlo desde el ecosistema:
Código
jupyter-lab ip=0.0.0.0Para facilitar el arranque de jupyter puede ser buena idea crear un script con las instrucciones necesarias y lanzarlo desde cron cada vez que arranque el usuario:
Scritp:
Código
#!/bin/bash
source jupyter/bin/activate
jupyter notebook –ip=0.0.0.0
#!/bin/bash
#! es un magistro especial que indica que la primera línea del archivo es el nombre del intérprete (en este caso, `/bin/bash`). Esto asegura que el script se ejecute con el entorno shell de Bash.
source jupyter/bin/activate
#! El comando source o “./” es un comando del shell que ejecuta otro script en el mismo entorno shell. “./jupyter/bin/activate” es el script que activa el entorno virtual. En este caso el directorio jupyter debe contener un entorno ya creado (por ejemplo el creado previamente con python3 -m venv jupyter). Al ejecutar esta línea, el entorno virtual se activa, y los comandos posteriores (como jupyter notebook) usarán las dependencias de este entorno virtual.
jupyter notebook –ip=0.0.0.0
#! jupyter notebook es el comando principal para iniciar el servidor de Jupyter Notebook. El parámetro `--ip=0.0.0.0` configura la dirección IP del servidor para que acepte conexiones desde cualquier dispositivo (no solo localhost). Esto es útil si se quiere acceder al cuaderno desde otro dispositivo o navegador remoto, o como en este curso a través de la máquina virtual.Línea de crontab para lanzar jupyter al arranque:
Código
@reboot /home/jupyter/startjupyter.sh
@reboot: Es un directivo especial en crontab que indica que el comando debe ejecutarse al momento en que el sistema inicia (durante el reinicio o el arranque). No necesita una hora ni un minuto, ya que solo se ejecuta una vez al inicio del sistema.
/home/jupyter/startjupyter.sh: Es la ruta completa del script Bash que se encuentra en este caso en /home/jupyter/. El nombre del archivo es startjupyter.sh. Este script contiene las instrucciones para iniciar Jupyter Notebook, activando su entorno virtual.Una vez arrancado jupyter, el usuario puede instalar los kernel que requiera, como por ejemplo BASH y R
2.2 Exploración preliminar de datos biológicos en R
La exploración preliminar de datos biológicos es un paso fundamental en cualquier análisis bioinformático, ya que permite entender la estructura y calidad de los datos antes de aplicar técnicas más avanzadas. En R, existen diversas herramientas y paquetes que facilitan esta tarea, especialmente cuando se trabaja con conjuntos de datos biológicos como secuencias de ADN, ARN o proteínas. A continuación, se describen los aspectos clave de esta exploración.
2.2.1 Manipulación de conjuntos de datos biológicos.
En biología computacional, los conjuntos de datos biológicos suelen ser grandes y complejos, por lo que es esencial saber manipularlos eficientemente. En R, paquetes como dplyr y tidyverse son ampliamente utilizados para la manipulación de datos. Estas herramientas permiten realizar operaciones como filtrado, selección de columnas, agrupación y resumen de datos. Por ejemplo, si se tiene un conjunto de datos que incluye información sobre genes (como nombres, longitudes y niveles de expresión), se pueden usar funciones como filter(), select() y summarize() para extraer información relevante y preparar los datos para análisis posteriores.
2.2.2 Importación y manejo de archivos de secuencias en formatos FASTA y FASTQ.
Los formatos FASTA y FASTQ son estándares en bioinformática para almacenar secuencias biológicas. En R, el paquete Biostrings (parte de Bioconductor) es especialmente útil para importar y manejar estos archivos. Para archivos FASTA, la función readDNAStringSet() permite cargar secuencias de ADN, mientras que readAAStringSet() se usa para secuencias de proteínas. En el caso de archivos FASTQ, que incluyen información de calidad, la función readFastq() del paquete ShortRead es la más adecuada. Una vez importadas, las secuencias pueden manipularse y analizarse utilizando funciones específicas de Biostrings, como width() para obtener la longitud de las secuencias o alphabetFrequency() para calcular la composición de bases.
Mas adelante en este curso se trabajará con alguna de estas librerías.
Es importante conocer que de igual manera que ocurre con BASH, la instalación de paquetes en R debe realizarse desde la consola de R, para ello, en el terminal de BASH escriba el comando:
R
Esto abrirá la consola de R:
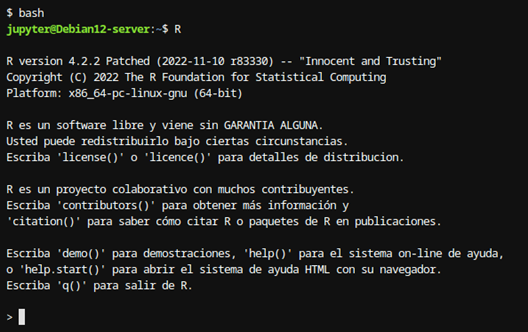
Figura 2: Consola R Esto mostrará el símbolo “>”, indicando que el prompt de R está listo para recibir instrucciones.
Ya dentro de la consola de R puede instalar nuevos paquetes, como por ejemplo BiocManager:
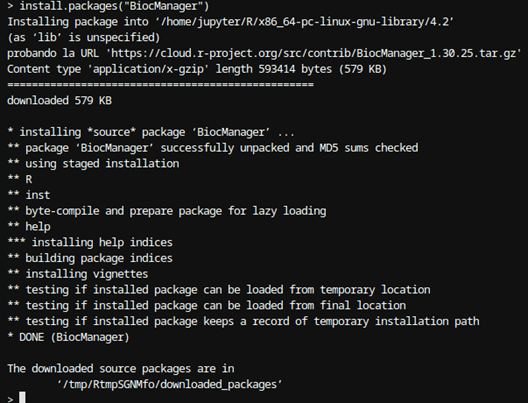
Figura 3: Instalación de BiocManager en R Una vez instalado BiocManager, a través de este, desde la misma consola de R, puede instalar BioString; BiocManager::install("Biostrings"):

Cuando termine el proceso le mostrará la siguiente pantalla, en la cual en este caso pregunta si desea actualizar algunos paquetes desactualizados:
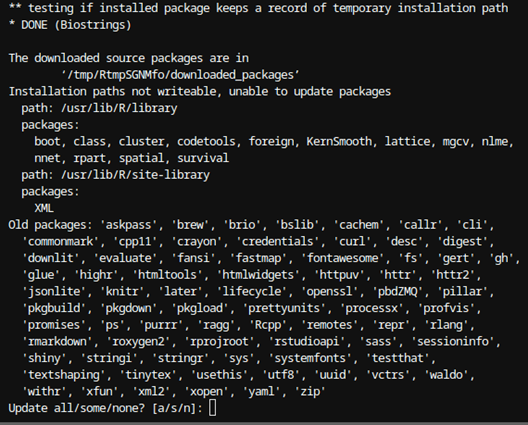
Figura 5: Fin de la instalación de BiocManager en R Si selecciona “a”, se actualizarán los paquetes mostrados, lo cual requerirá algunos minutos hasta descargar, compilar e instalarlos. Dado que el ecosistema de R tiene muchas librerías, es posible que al actualizar ese volumen de ellas tenga problemas de dependencias, por lo que se recomienda no hacerlo en este caso.
Para salir de la consola de R, digite quit()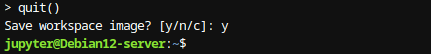
Figura 6: Salir de consola Una vez que los paquetes estén instalados, para cargar las librerías en R, se utilizan funciones como library() o require(). La función library(nombre_del_paquete) carga el paquete especificado, mientras que require(Nombre_del_paquete) realiza una carga condicional del paquete solo si no está cargado previamente. Por ejemplo:
Código
library(ggplot2) # Carga el paquete ggplot2Si el paquete no se encuentra en su sistema, R mostrará un mensaje de error. Puede instalarlo usando:
Código
install.packages("ggplot2")Es importante verificar la versión del paquete y asegurarse de que cumpla con las dependencias. Si necesita cargar múltiples paquetes, puede incluirlos en una sola línea o usar un script de inicio (Rprofile), sin embargo esta opción puede enmascarar y dificultar el detectar posibles problemas de dependencias durante el proceso de instalación de esos múltiples paquetes, en cuyo caso deberá realizarlos en grupos mas pequeños hasta detectar el problema.
2.2.3 Exploración de datos de secuencias: estadísticas básicas (longitud de secuencias, composición de bases).
Una vez importadas las secuencias, es importante realizar un análisis exploratorio para entender sus características. En R, se pueden calcular estadísticas básicas como la longitud de las secuencias y la composición de bases. Por ejemplo, la función width() del paquete Biostrings permite obtener la longitud de cada secuencia, mientras que alphabetFrequency() calcula la frecuencia de cada nucleótido (A, T, C, G) o aminoácido en el caso de proteínas. Estas estadísticas son útiles para identificar patrones, como sesgos en la composición de bases o secuencias anómalas que podrían requerir limpieza o filtrado antes de proceder con análisis más avanzados.
2.2.4 Prácticas sobre cómo obtener y limpiar datos biológicos para su análisis.
La obtención y limpieza de datos biológicos es un paso crítico en cualquier proyecto de bioinformática. En R, se pueden utilizar paquetes como rentrez para acceder a bases de datos públicas como GenBank y descargar secuencias de interés. Una vez obtenidos los datos, es común encontrar problemas como secuencias incompletas, baja calidad (en el caso de FASTQ) o formatos inconsistentes. Para limpiar los datos, se pueden aplicar técnicas como el filtrado de secuencias cortas o de baja calidad, la corrección de errores y la normalización de formatos. Herramientas como ShortRead y Biostrings ofrecen funciones para realizar estas tareas de manera eficiente. Por ejemplo, se puede usar trimTails() para recortar secuencias de baja calidad en archivos FASTQ o unique() para eliminar secuencias duplicadas.
Resumen
Esta parte del curso introduce al investigador a trabajar en biología computacional utilizando R y Python, destacando las diferencias y complementariedades entre ambos lenguajes. Además, se enfoca en la exploración y preparación de datos biológicos, un paso esencial antes de realizar análisis más avanzados. La combinación de herramientas y paquetes especializados en R y Python permite a los investigadores abordar problemas biológicos de manera eficiente y reproducible. En esta primera sección se cubre una introducción a R en la biología computacional y a través del paquete Biostring se trabajó con secuencias de ADN para obtener datos de estas.