Diagrama de temas
-
Ejemplos de uso avanzado de Bioconductor
-
3.3. Ejemplos de uso avanzado de Bioconductor
3.3.1. Implementación de tareas avanzadas con Bioconductor
Bioconductor es una herramienta poderosa para el análisis de datos genómicos y biológicos. Entre las tareas avanzadas que se pueden implementar se encuentran:
Anotación de secuencias: Utilizando paquetes como AnnotationDbi y org.Hs.eg.db, es posible anotar secuencias de ADN o ARN con información biológica relevante, como genes, transcripciones y funciones biológicas. Por ejemplo, se pueden mapear identificadores de genes (como ENSEMBL o Entrez) a nombres de genes y descripciones funcionales.
Análisis de datos de expresión génica: Con paquetes como limma y DESeq2, se pueden realizar análisis diferenciales de expresión génica a partir de datos de microarrays o RNA-seq. Esto incluye la normalización de datos, identificación de genes diferencialmente expresados y visualización de resultados mediante heatmaps o gráficos de dispersión.
3.3.2. Ejercicios prácticos
Ejercicio con BIOCONDUCTOR
A continuación, se desarrolla un ejercicio utilizando Bioconductor en R.
Este ejercicio cubre desde la carga de datos hasta el , incluyendo la visualización de resultados. Utilizaremos datos de RNA-seq como ejemplo.
Ejercicio: Análisis de Expresión Diferencial con Bioconductor
Objetivo
Realizar un análisis de expresión diferencial (DEA) a partir de datos de RNA-seq para identificar genes que están regulados diferencialmente entre dos condiciones (por ejemplo, tratamiento vs control).
Pasos del Ejercicio
- Instalación de paquetes necesarios.
- Carga de datos de expresión génica.
- Preprocesamiento y normalización de datos.
- Análisis de expresión diferencial.
- Visualización de resultados.
- Interpretación y generación de reportes.
Aprende más
Para desarrollar este ejercicio ejecute el cuaderno de trabajo 11 ¡Accede aquí!
A continuación, se desarrolla un ejercicio utilizando Bioconductor en R.
- Instalación de paquetes necesarios (En caso de requerirse)
- Carga de datos de expresión génica
- Origen de los datos: Los datos en airway provienen de un estudio publicado en 2013 por Himes et al. en la revista PLoS ONE. El estudio analizó el efecto de la dexametasona (un glucocorticoide sintético) en células de las vías respiratorias humanas (airway smooth muscle cells).
- Tipo de datos: Contiene datos de RNA-seq, que incluyen conteos de lecturas (reads) mapeados a genes para varias muestras.
- Formato: Los datos están almacenados en un objeto de tipo SummarizedExperiment, que es una estructura de datos común en Bioconductor para almacenar y manipular datos genómicos.
- Conteos de lecturas (counts):
- Matriz de conteos de lecturas mapeadas a genes para cada muestra.
- Las filas representan genes (identificados por ENSEMBL IDs).
- Las columnas representan muestras (4 tratadas con dexametasona y 4 no tratadas).
- Metadatos de las muestras (colData):
- Información sobre las muestras, como:
- SampleName: Identificador de la muestra.
- cell: Línea celular.
- dex: Condición de tratamiento (tratado con dexametasona o no tratado).
- albut: Indicador de uso de albuterol (un broncodilatador).
- Run: Identificador de la corrida de secuenciación.
- Metadatos de los genes (rowData):
- Información sobre los genes, como sus identificadores (ENSEMBL IDs).
- assays: Contiene la matriz de conteos de lecturas.
- colData: Contiene los metadatos de las muestras.
- rowData: Contiene los metadatos de los genes
- metadata: Información adicional sobre el experimento.
Primero, instalamos los paquetes de Bioconductor necesarios para este análisis:
Ejecutar desde la consola de R:
Código
############ if (!require("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(c("DESeq2", "airway", "pheatmap", "ggplot2", "AnnotationDbi", "org.Hs.eg.db")) ############
Utilizaremos el paquete airway, que contiene un conjunto de datos de RNA-seq de un experimento con células de las vías respiratorias humanas tratadas con dexametasona.
La librería airway es un paquete de datos en Bioconductor que proporciona un conjunto de datos de RNA-seq (secuenciación de ARN) preprocesado y listo para usar. Estos datos provienen de un estudio científico real y se utilizan comúnmente como un ejemplo estándar en tutoriales y análisis de bioinformática, especialmente para demostrar el uso de herramientas como DESeq2 para el análisis de expresión diferencial.
El objeto airway incluye:
El objeto airway es un SummarizedExperiment, que tiene la siguiente estructura:
Una matriz de conteos de lecturas mapeadas a genes (filas: genes, columnas: muestras).
Información sobre las muestras, como la condición de tratamiento (dex: dexametasona) y la línea celular.
Identificadores de genes (ENSEMBL IDs).
El conjunto de datos airway se utiliza principalmente para demostrar el flujo de trabajo de análisis de RNA-seq
- 1. Carga de datos
- Conversión a objeto DESeqDataSet
- Los conteos de lecturas.
- Los metadatos de las muestras.
- El diseño experimental para el análisis.
- Filtrado de genes con baja expresión
- Como criterio de filtrado se conservan los genes que tienen al menos 10 lecturas en total en todas las muestras combinadas.
- El objeto dds ahora contiene solo los genes que cumplen con el criterio de expresión mínima.
- Esto reduce el número de genes y mejora la precisión del análisis.
- Normalización y análisis de expresión diferencial
- Estimación de factores de tamaño: Ajusta las diferencias en la profundidad de secuenciación entre muestras.
- Dispersión de genes: Estima la variabilidad biológica entre muestras.
- Ajuste del modelo: Compara las condiciones de tratamiento para identificar genes diferencialmente expresados.
- Extracción de resultados
- log2FoldChange: Cambio en la expresión (en escala logarítmica) entre las condiciones.
- pvalue: Valor p sin ajustar.
- padj: Valor p ajustado (corregido por múltiples pruebas).
- baseMean: Expresión media normalizada del gen.
- log2FoldChange > 0: El gen está sobreexpresado en la condición tratada.
- log2FoldChange < 0: El gen está subexpresado en la condición tratada.
- padj < 0.05: El gen se considera significativamente diferencialmente expresado.
- Visualización de resultados
- MA plot (Mean of Normalized Counts vs Log Fold Change), y;
- Heatmap con dendrograma (muestras SRRXXXXXXX vs genes ENSG00000XXXXXX).
Código
############ library(DESeq2) library(airway) # Cargar el conjunto de datos data("airway") se <- airway ############
Exploración de los datos:
Código
############ # Ver el objeto airway ############ # Ver los conteos de lecturas head(assay(airway)) tail(assay(airway)) ############ # Ver metadatos de las muestras colData(airway) ############ # Ver metadatos de los genes rowData(airway)
Análisis de expresión diferencial:
El paquete airway se usa comúnmente con DESeq2 para realizar análisis de expresión diferencial entre las muestras tratadas y no tratadas con dexametasona.
El objetivo es convertir el objeto airway en un DESeqDataSet, que es el formato requerido por el paquete DESeq2 para realizar el análisis de expresión diferencial.
Diseño experimental: El argumento “design = ~dex” especifica que queremos comparar las muestras en función de la condición de tratamiento (dex: dexametasona).
El objeto dds contendrá:
Código
######## library(DESeq2) # Convertir a objeto DESeqDataSet dds <- DESeqDataSet(airway, design = ~ dex)
Tiene por objeto eliminar genes con baja expresión, ya que no proporcionan información útil para el análisis.
Código
######## # Filtrar genes con baja expresión keep <- rowSums(counts(dds)) >= 10 dds <- dds[keep, ] ########
En este paso se busca normalizar los datos y realizar el análisis de expresión diferencial.
Pasos que realiza DESeq:
El objeto dds ahora contendrá los resultados del modelo estadístico.
Código
######## # Normalización y análisis dds <- DESeq(dds) ########
En este punto se extraen los resultados del análisis de expresión diferencial.
Contraste: Especificamos que queremos comparar las muestras tratadas con dexametasona (trt) frente a las no tratadas (untrt).
El objeto res contiene:
Aquí se puede interpretar que:
Código
######## res <- results(dds, contrast = c("dex", "trt", "untrt")) ########
Se pueden generar gráficos como heatmaps, MA plots y PCA para visualizar los resultados.
Código
######## # MA plot plotMA(res) # Heatmap de los genes más significativos library(pheatmap) top_genes <- head(order(res$padj), 20) pheatmap(assay(dds)[top_genes, ], scale = "row") ########
Como resultado se obtienen los gráficos:
Ambos son herramientas visuales comunes en el análisis de expresión diferencial y proporcionan información complementaria sobre los resultados.
Gráfico MA Plot (Mean of Normalized Counts vs Log Fold Change)
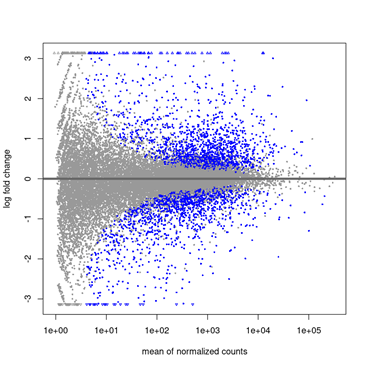
Figura 1: MA Plot (Mean of Normalized Counts vs Log Fold Change) Este gráfico muestra en el eje X (Mean of Normalized Counts) la expresión media normalizada de un gen en todas las muestras (en escala logarítmica), mientras que en el eje Y (Log Fold Change) el cambio en la expresión de un gen entre dos condiciones (en escala logarítmica base 2).
Puntos: Cada punto representa un gen.
Línea horizontal en Y=0: Indica que no hay cambio en la expresión entre las condiciones.
Interpretación:
- Puntos cerca de Y=0:
- Genes con poco o ningún cambio en la expresión entre las condiciones.
- Estos genes no son de interés en el análisis de expresión diferencial.
- Puntos por encima de Y=0:
- Genes con log2FoldChange > 0 (sobreexpresados en la condición tratada).
- Cuanto más alejados estén de Y=0, mayor es el cambio en la expresión.
- Puntos por debajo de Y=0:
- Genes con log2FoldChange < 0 (subexpresados en la condición tratada).
- Cuanto más alejados estén de Y=0, mayor es el cambio en la expresión.
- Puntos coloreados (generalmente rojos):
- Representan genes significativamente diferencialmente expresados (con un padj < 0.05).
- Estos genes son los más relevantes para el análisis.
- Distribución de puntos:
- Si la mayoría de los puntos están cerca de Y=0, significa que la mayoría de los genes no cambian significativamente entre las condiciones.
- Si hay muchos puntos alejados de Y=0 (especialmente los coloreados), indica que hay muchos genes diferencialmente expresados.
Si un gen tiene un log2FoldChange = 2 y está coloreado en rojo, significa que está significativamente sobreexpresado en la condición tratada (4 veces más expresado).
Si un gen tiene un log2FoldChange = -1.5 y está coloreado en rojo, significa que está significativamente subexpresado en la condición tratada (aproximadamente 2.83 veces menos expresado).
Gráfico Heatmap con Dendrograma (Muestras SRRXXXXXXX vs Genes ENSG00000XXXXXX)
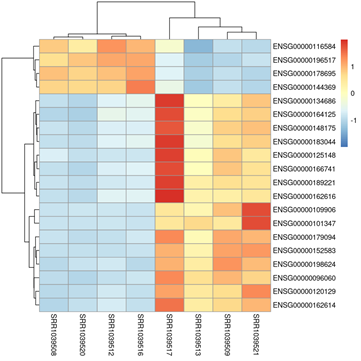
Figura 2: Heatmap con Dendrograma (Muestras SRRXXXXXXX vs Genes ENSG00000XXXXXX) Este gráfico de mapa de calor (heatmap) con dendrograma es una representación gráfica en la que los valores de expresión de los genes se representan con colores (rojo para sobreexpresión y azul para subexpresión).
El eje Y (filas) muestra los genes (identificados por ENSEMBL IDs, como ENSG00000XXXXXX), mientras que el eje X (columnas) presenta las muestras (identificadas por códigos como SRRXXXXXXX).
El dendrograma es un árbol jerárquico que agrupa muestras o genes basándose en su similitud en los patrones de expresión.
- Colores en el heatmap:
- Rojo: Indica sobreexpresión del gen en esa muestra.
- Azul: Indica subexpresión del gen en esa muestra.
- Blanco o tonos claros: Indica expresión media o sin cambios.
- Agrupación de muestras (dendrograma superior):
- Muestras que se agrupan juntas tienen patrones de expresión similares.
- Si las muestras de la misma condición (por ejemplo, tratadas vs no tratadas) se agrupan juntas, sugiere que el tratamiento tiene un efecto claro en la expresión génica.
- Agrupación de genes (dendrograma lateral):
- Genes que se agrupan juntos tienen patrones de expresión similares en todas las muestras.
- Esto puede indicar que los genes están co-regulados o tienen funciones biológicas relacionadas.
- Genes diferencialmente expresados:
- Los genes más relevantes son aquellos que muestran un patrón claro de sobreexpresión o subexpresión en las muestras de una condición específica.
Si las muestras SRRXXXXXXX de la condición tratada se agrupan juntas y muestran un patrón de sobreexpresión (rojo) para un conjunto de genes, esto sugiere que esos genes están regulados positivamente por el tratamiento.
Si un grupo de genes ENSG00000XXXXXX se agrupa y muestra subexpresión (azul) en las muestras tratadas, esto sugiere que esos genes están regulados negativamente por el tratamiento.
3.3.3. Aplicación de funciones estadísticas y generación de reportes
Bioconductor permite aplicar funciones estadísticas avanzadas para el análisis de datos biológicos. Por ejemplo, se pueden realizar análisis de enriquecimiento funcional con el paquete clusterProfiler para identificar rutas biológicas significativas. Además, se pueden generar reportes automatizados utilizando knitr y rmarkdown, integrando código, resultados y visualizaciones en un documento final.
En el ejercicio previo y el cuaderno de trabajo 11 se muestran ejemplos de reportes y visualizaciones.
3.4. Ejemplos de uso avanzado de BioPython
3.4.1. Implementación de análisis avanzados con BioPython
BioPython es una biblioteca de Python ampliamente utilizada en bioinformática. Entre sus funcionalidades avanzadas se encuentran:
- Alineamiento de secuencias: Utilizando el módulo Bio.Align, se pueden realizar alineamientos locales y globales de secuencias con algoritmos como Needleman-Wunsch y Smith-Waterman. También se pueden realizar alineamientos múltiples con herramientas como ClustalW o MUSCLE.
- Búsqueda de motivos en secuencias: Con el módulo Bio.motifs, se pueden buscar motivos específicos en secuencias de ADN o ARN, lo que es útil para identificar sitios de unión de factores de transcripción o secuencias reguladoras.
En la clase 7 se ha desarrollado el cuaderno 10, donde se desarrollaron ejercicios con Biopython.
3.4.2. Ejercicios prácticos
Resolución de dependencias
Resolver dependencias es un paso crucial para garantizar que los proyectos en R y Python funcionen correctamente. Estas dependencias pueden incluir tanto librerías del sistema operativo (por ejemplo, en Debian) como paquetes o módulos de R y Python. A continuación, se describe el proceso para resolver estas dependencias en ambos lenguajes.
Ejecute el cuaderno:
Aprende más
Para desarrollar el ejercicio puedes descargar el siguiente cuaderno de trabajo 12 ¡Accede aquí!
- Cuaderno12_Librerias_Python_adicionales_Apellido_Nombre.ipynb
Para comenzar actualizaremos el sistema operativo Debian®:
Código
sudo apt update sudo apt upgrade
Al finalizar le preguntará dónde escribir el GRUB; seleccione /dev/sda
Figura 3: Selección de unidad para escribir el GRUB Reinicie la máquina para verificar que todo ha ido bien:
Código
sudo reboot
PIP
es el gestor de paquetes estándar para Python, utilizado para instalar, actualizar y gestionar bibliotecas y dependencias de software escritas en este lenguaje. Paera actualizar este gestor ejecute:
Código
pip install --upgrade pip
Es posible que se le muestre la advertencia “DEPRECATION: Loading egg at /home/jupyter/jupyter/lib/python3.11/site-packages/SeqUtils-1.0.11-py3.11.egg is deprecated. pip 25.1 will enforce this behaviour change. A possible replacement is to use pip for package installation. Discussion can be found at https://github.com/pypa/pip/issues/12330”
Esto muestra que la librería SeqUtils quedará obsoleta en futuras versiones de este ecosistema. Si no quiere recibir esta advertencia cada vez que use pip, puede desinstalarla con:
Código
pip uninstall SeqUtils -y #Librería pandas para el manejo de dataframes pip install pandas #Librerías para graficación: pip install matplotlib pip install seaborn pip install graphviz pip install pydot #Librería para análisis estadístico pip install statsmodels pip install pingouin #Librería para interactuar con las rutas de los sistemas operativos pip install pathlib #Librerías para biología computacional pip install biopython #Librerías para aprendizaje automático pip install scikit-learn pip install mlxtend #openpyxl para leer y escribir archivos Excel (R) 2010 pip install openpyxl pip install xlrd #Herramienta para el procesamiento de lenguaje natural pip install nltk #Herramienta para asociación (Aprendizaje no supervisado) pip install apyori #Herramienta para Big Data con Spark pip install pyspark #Exportar los cuadernos a PDF vía LaTEX #Abra un terminal y ejecute las siguientes instrucciones: sudo apt-get install texlive-xetex sudo apt-get install texlive-fonts-recommended
Instalación de librerías adicionales en R
Las siquientes librerías implementan herramientas para la capa de visualización en R por lo que de acuerdo a su necesidad se recomienda su instalación.
Aprende más
Para ello ejecute el cuaderno de trabajo 13 ¡Accede aquí!
Este cuaderno tiene como objetivo:
- Instalar librerías adicionales en R
- Instalar paquetes en R desde repositorios específicos
- Instalar paquetes en R desde fuentes exrernas como github
- Resolver problemas de dependencias con librerías o paquetes de sistema operativo (Debian®)
Resumen
En este curso, se exploraron herramientas BASH aplicadas a la biología computacional, así como herramientas como Bioconductor y BioPython para el análisis de datos biológicos. En la sección de Bioconductor, se abordaron tareas como el análisis de expresión génica, utilizando paquetes como DESeq2. Se realizaron ejercicios prácticos, como la conversión de secuencias de ADN a ARN y el análisis de variantes genéticas, además de aplicar funciones estadísticas para interpretar datos biológicos y generar reportes automatizados. Estos ejercicios permitieron comprender cómo manipular y analizar datos genómicos de manera eficiente.
Por otro lado, en la sección de BioPython a través de ejercicios prácticos, se analizaron archivos de secuencias en formatos como FASTA y GenBank, y se realizaron cálculos sobre la composición de bases nitrogenadas, se destacó la integración de BioPython con otras bibliotecas de procesamiento de datos, como pandas y matplotlib, para visualizar resultados y realizar análisis más complejos.
Finalmente, el curso combinó teoría y práctica, proporcionando una visión integral de cómo utilizar Bioconductor y BioPython en el análisis de datos biológicos. Desde la manipulación de secuencias y la identificación de genes diferencialmente expresados hasta la generación de reportes y visualizaciones.